






")


")












Sổ tay chất lượng đăng ký vắc xin sinh phẩm y tế
BỘ Y TẾ
CỤC QUẢN LÝ DƯỢC
SỔ TAY CHẤT LƯỢNG
ĐĂNG KÝ VẮC XIN – SINH PHẨM Y TẾ
Mã số : QM
Lần ban hành : 01
Ngày ban hành : 05.07.2010
| Biên soạn | Xem xét | Phê duyệt | |
| Họ tên | Đỗ Minh Hùng | Phạm Hồng Châu | Trương Quốc Cường |
| Ký tên | (ĐÃ KÍ) | (ĐÃ KÍ) | (ĐÃ KÍ) |
Theo dõi sửa đổi tài liệu
| TT | Vị trí | Nội dung sửa đổi | Ngày sửa đổi |
CHƯƠNG 1: MỤC LỤC
| Nội dung | Trang | |
| Chương 1 | MỤC LỤC | 2 |
| Chương 2 | CHÍNH SÁCH CHẤT LƯỢNG | 4 |
| Chương 3 | GIỚI THIỆU SỔ TAY CHẤT LƯỢNG | 5 |
| Chương 4 | CƠ CẤU TỔ CHỨC ĐĂNG KÝ VẮC XIN – SINH PHẨM Y TẾ | 6 |
| 4.1. | Sơ đồ tổ chức | 6 |
| 4.2 | Cục Quản lý dược | 6 |
| 4.3 | Hội đồng xét duyệt | 8 |
| 4.4 | Hội đồng chuyên gia thẩm định | 8 |
| 4.5 | Nguồn nhân sự | 9 |
| Chương 5 | QUÁ TRÌNH ĐĂNG KÝ VẮC XIN – SINH PHẨM Y TẾ | 11 |
| 5.1 | Sơ đồ quy trình | 11 |
| 5.2 | Qui định đăng ký | 11 |
| 5.3 | Nộp đơn đề nghị đăng ký | 12 |
| 5.4 | Xem xét nguồn lực | 12 |
| 5.5 | Nguyên tắc hoạt động thẩm định | 12 |
| 5.6 | Thực hiện quy trình | 12 |
| 5.7 | Đánh giá thực tế | 12 |
| 5.8 | Yêu cầu xem xét khiếu nại | 13 |
| 5.9 | Thực hiện quy trình nhanh | 14 |
| Chương 6 | ĐẢM BẢO CHẤT LƯỢNG ĐĂNG KÝ VẮC XIN – SINH PHẨM Y TẾ | 15 |
| 6.1 | Quy định chung | 15 |
| 6.2 | Hệ thống quản lý | 15 |
| 6.3 | Kiểm soát tài liệu | 15 |
| 6.4 | Sự không phù hợp và hành động khắc phục | 15 |
| 6.5 | Hành động phòng ngừa | 15 |
| 6.6 | Đánh giá nội bộ | 16 |
| 6.7 | Xem xét của Lãnh đạo | 16 |
| 6.8 | Phàn nàn | 16 |
CHƯƠNG 2: CHÍNH SÁCH CHẤT LƯỢNG VỀ ĐĂNG KÝ VẮC XIN – SINH PHẨM Y TẾ
Cục Quản lý dược thực hiện các chính sách:
Góp phần nâng cao năng lực của Quản lý Nhà nước về công việc đăng ký lưu hành vắc xin – sinh phẩm y tế (bao gồm tổ chức nhận hồ sơ, thẩm định, xét duyệt, cấp số đăng ký), tạo sự tin cậy của nhân dân vào công tác cấp số đăng ký lưu hành đối với vắc xin – sinh phẩm y tế; phấn đấu cho mục tiêu “chuẩn mực trong đăng ký lưu hành vắc xin – sinh phẩm y tế”.
Xây dựng và áp dụng hệ thống quản lý chất lượng phù hợp với tiêu chuẩn ISO và hoà hợp quốc tế.
Coi trọng nguồn nhân lực, tạo điều kiện và cơ hội cho việc đào tạo, nâng cao trình độ, học hỏi kinh nghiệm phục vụ cho công việc đăng ký vắc xin – sinh phẩm y tế.
Tôn trọng, bình đẳng với mọi đối tượng doanh nghiệp. Không có bất cứ một điều kiện hoặc áp lực nào dẫn đến phân biệt đối xử giữa các doanh nghiệp đăng ký lưu hành vắc xin – sinh phẩm y tế. Thực hiện phương châm hành động là khách quan – trung thực – đúng đắn – kịp thời.
CHƯƠNG 3: GIỚI THIỆU SỔ TAY CHẤT LƯỢNG
3.1. Mục đích của cuốn sổ tay chất lượng (STCL) là công bố các chính sách và mục tiêu chất lượng của Cục Quản lý dược về công tác đăng ký vắc xin – sinh phẩm y tế tại Việt Nam nhằm thoả mãn yêu cầu của tiêu chuẩn ISO và các quy định pháp quy có liên quan.
3.2. Cấu trúc STCL
STCL gồm 6 chương chính là: Mục lục; Chính sách chất lượng; Giới thiệu STCL; Cơ cấu tổ chức; Quá trình đăng ký; Đảm bảo chất lượng.
3.3. Phê duyệt: STCL được Cục Trưởng Cục Quản lý dược phê duyệt.
3.4. Cơ sở Pháp lý của sổ tay chất lượng:
– Quyết định số 53/2008/QĐ-BYT ngày 30/12/2008 của Bộ Trưởng Bộ Y tế quy định chức năng, nhiệm vụ của Cục Quản lý Dược.
– Thông tư số 22/2009/TT-BYT của Bộ Y tế ngày 24/11/2009 quy định việc đăng ký thuốc.
3.5. Tính pháp lý và phạm vi hiệu lực của STCL
STCL có hiệu lực áp dụng kể từ ngày ký ban hành.
STCL được ban hành, quản lý và áp dụng tại Cục Quản lý dược trong các hoạt động liên quan đến đăng ký vắc xin – sinh phẩm y tế.
CHƯƠNG 4: CƠ CẤU TỔ CHỨC VỀ ĐĂNG KÝ VẮC XIN – SPYT
Mục tiêu:
– Đảm bảo chất lượng công tác đăng ký vắc xin- sinh phẩm y tế.
– Đảm bảo việc truy tìm các sai xót, tồn tại, bất cập để khắc phục trong quá trình đăng ký vắc xin, sinh phẩm y tế.
1- Sơ đồ tổ chức: liên quan trực tiếp đến hoạt động đăng ký
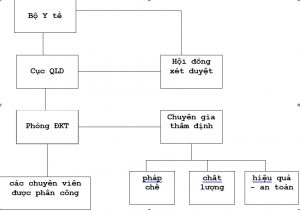
2- Cục Quản lý dược:
2.1. Trách nhiệm pháp lý
Cục Quản lý dược là đơn vị thuộc Bộ Y tế được quy định chức năng, nhiệm vụ theo Quyết định số 53/2008/QĐ-BYT ngày 30/12/2008 của Bộ Trưởng Bộ Y tế.
Cục Quản lý dược phân định rõ trách nhiệm và quyền hạn của từng bộ phận, cá nhân trong việc điều hành, quản lý liên quan đến quá trình đăng ký vắc xin – sinh phẩm y tế. Quá trình đó được giám sát chặt chẽ, đúng thủ tục và kịp thời phát hiện, khắc phục những điều không phù hợp, đảm bảo duy trì và kiểm soát hệ thống quản lý;
Mọi cá nhân được giao nhiệm vụ tham gia vào hoạt động đánh giá, thẩm định đăng ký lưu hành vắc xin phải tuân thủ quy định về trách nhiệm, quản lý của Cục QLD;
2.2. Cơ cấu tổ chức
2.2.1. Cục Trưởng Cục Quản lý dược
– Đại diện cao nhất của Cục Quản lý dược trong các mối quan hệ hợp tác song phương và hoạt động của các tổ chức quốc tế và khu vực về đăng ký lưu hành vắc xin – sinh phẩm y tế;
– Ban hành chính sách chất lượng, thường xuyên giám sát việc triển khai chính sách và thủ tục về đăng ký vắc xin và sinh phẩm y tế.
– Duyệt và ký các văn bản thuộc hệ thống tài liệu liên quan đến đảm bảo chất lượng công tác đăng ký lưu hành vắc xin – sinh phẩm y tế;
– Định kỳ 1 năm 1 lần xem xét và phê chuẩn việc cải tiến hệ thống quản lý đăng ký vắc xin – sinh phẩm y tế;
– Quy định trách nhiệm, quyền hạn, phân công công việc trong hoạt động đăng ký vắc xin – sinh phẩm y tế, duyệt kế hoạch và theo dõi việc thực hiện.
2.2.2- Phòng Đăng ký thuốc:
Phòng Đăng ký thuốc là một phòng chuyên môn của Cục Quản lý dược có nhiệm vụ tham mưu cho Lãnh đạo Cục QLD giải quyết các công việc liên quan đế đăng ký thuốc, vắc xin – sinh phẩm y tế.
2.2.2.1- Trưởng Phòng Đăng ký thuốc
Là người chịu trách nhiệm trước Cục Trưởng Cục Quản lý dược trong hoạt động tác nghiệp được phân công với các nhiệm vụ, quyền hạn sau:
– Giúp Cục Trưởng trong việc điều hành các hoạt động tác nghiệp được phân công;
– Tổ chức phân công và điều hành việc đăng ký vắc xin – sinh phẩm y tế theo lĩnh vực được phân công;
2.2.2.2- Nhóm theo dõi đăng ký lưu hành vắc xin – sinh phẩm y tế (thuộc Phòng ĐKT)
Nhóm theo dõi đăng ký lưu hành vắc xin – sinh phẩm y tế thực hiện các công việc theo sự phân công của Trưởng Phòng Đăng ký thuốc làm đầu mối hoạt động đăng ký vắc xin – sinh phẩm y tế có các nhiệm vụ, quyền hạn sau:
– Chuẩn bị các vấn đề liên quan đến tổ chức để các chuyên gia thẩm định hồ sơ và tổ chức các cuộc họp Hội đồng xét duyệt;
– Theo dõi việc tuân thủ các thủ tục, hướng dẫn, biểu mẫu đã quy định, kiểm tra tính pháp lý của kết quả thẩm định;
– Chuẩn bị các văn bản liên quan đến việc cấp số đăng ký (quyết định cấp số đăng ký);
– Quản lý hồ sơ đăng ký lưu hành vắc xin – sinh phẩm y tế
– Theo dõi hoạt động thẩm định hồ sơ của các chuyên gia thẩm định;
– Lập báo cáo hoạt động và quản lý hồ sơ về đăng ký vắc xin – sinh phẩm y tế.
- Nhóm chuyên gia thẩm định.
Nhóm chuyên gia thẩm định do Cục Trưởng Cục Quản lý dược thành lập. Thành viên của nhóm chuyên gia thẩm định là những người có trình độ, kiến thức, kinh nghiệm và năng lực trong các lĩnh vực thẩm định (Pháp chế; Quy trình sản xuất – chất lượng; Dược lý – lâm sàng).
Chuyên gia thẩm định có chức năng, nhiệm vụ sau:
3.1- Chức năng, nhiệm vụ:
Chuyên gia thẩm định có nhiệm vụ tư vấn cho Cục Quản lý Dược trong việc thẩm định hồ sơ đăng ký thuốc và đề xuất việc cấp số đăng ký hoặc bổ sung hoặc không cấp số đăng ký thuốc.
Chuyên gia thẩm định hoạt động trên nguyên tắc: các ý kiến góp ý, và/hoặc đề xuất của chuyên gia thẩm định phải đảm bảo căn cứ pháp lý, cơ sở khoa học và phải được thể hiện trong Biên bản thẩm định hồ sơ đăng ký thuốc. Chuyên gia thẩm định chịu trách nhiệm trước Cục trưởng Cục Quản lý Dược về các nội dung/ý kiến tư vấn và đề xuất liên quan đến công tác thẩm định hồ sơ đăng ký thuốc
Thực hiện các nhiệm vụ cụ thể trong từng lĩnh vực thẩm định như sau:
3.2.1- Nhóm 1: Thẩm định hồ sơ về hành chính và thông tin sản phẩm:
a) Mục đích:
– Đảm bảo tính pháp lý của toàn bộ hồ sơ, các hồ sơ công ty nộp là phù hợp với các quy định hiện hành về hình thức, về tính hợp pháp;
– Phần Hồ sơ pháp lý phản ánh đúng bản chất của thuốc, các thông tin chính xác
– Xác định các vấn đề cần quan tâm và đề xuất biện pháp để đảm bảo có kết luận chính xác trong toàn bộ quá trình thẩm định.
b) Nội dung thẩm định: Thẩm định phần hồ sơ pháp lý và tính pháp lý của các phần hồ sơ kỹ thuật; Nhãn thuốc; Sở hữu trí tuệ và nhãn hiệu hàng hoá.
c) Căn cứ để thẩm định:
– Thông tư đăng ký thuốc hiện hành
– Hướng dẫn đăng ký thuốc
– Các quy định hiện hành có liên quan đến tính hợp pháp của văn bản: công chứng, chứng thực, …..
3.2.2- Nhóm 2: Thẩm định hồ sơ về chất lượng:
a) Mục đích: đảm bảo chất lượng thuốc, phương pháp kiểm nghiệm phù hợp
b) Nội dung thẩm định:
Thuộc phần hồ sơ chất lượng trong bộ hồ sơ đăng ký.
c) Căn cứ và tài liệu tham khảo:
– Thông tư số 22/2009/TT-BYT quy định việc đăng ký thuốc.
– Dược Điển Việt Nam; Dược điển các nước
– Các Nguyên tắc thực hành tốt sản xuất thuốc; Thực hành tốt Phòng thí nghiệm; Thực hành tốt bảo quản thuốc;
– Các hướng dẫn của Việt Nam và quốc tế liên quan.
3.2.3- Nhóm 3: Thẩm định hồ sơ về an toàn và hiệu quả
a) Mục đích: đảm bảo thuốc được phép lưu hành an toàn và hiệu quả đối với bệnh nhân là người Việt Nam;
b) Nội dung thẩm định: Các phần hồ sơ an toàn, hiệu quả trong bộ hồ sơ đăng ký.
c) Các căn cứ và tài liệu tham khảo:
– Quyết định số 01/2007/TT-BYT về thử thuốc trên lâm sàng;
– Thông tư về đăng ký thuốc;
– Hướng dẫn thực hành tốt thử thuốc trên lâm sàng;
– Các tài liệu tham khảo chính thức: Martindal; Dược điển Anh, Mỹ, Châu Âu; Kết quả thẩm định lâm sàng của Cơ quan quản lý dược các nước USFDA, EMEA, TGA
– Các tài liệu liên quan khác.
4- Hội đồng xét duyệt hồ sơ đăng ký lưu hành vắc xin – sinh phẩm y tế
Hội đồng dược thành lập theo Quyết định số 1893/QĐ-BYT, ngày 28/5/2007 của Bộ Trưởng Bộ Y tế. Hội đồng bao gồm 16 thành viên là các chuyên gia, nhà khoa học đầu ngành về vắc xin – sinh phẩm y tế từ các Viện nghiên cứu, bệnh viện, các đơn vị chuyên môn, quản lý thuộc Bộ Y tế có nhiệm vụ tư vấn cho Bộ trưởng Bộ Y tế về các vấn đề về đăng ký vắc xin – sinh phẩm y tế.
5- Phát triển nguồn nhân lực:
5.1. Nguồn nhân lực:
a) Nhân lực cán bộ của Cục Quản lý Dược liên quan đến đăng ký vắc xin – sinh phẩm y tế
Cục Quản lý dược phải duy trì một đội ngũ nhân viên đủ năng lực, đã được đào tạo, có kinh nghiệm chuyên môn, nghiệp vụ thích hợp để thực hiện các công việc được giao. Mỗi cán bộ phải được đào tạo theo hướng chuyên nghiệp. Cán bộ tham gia vào hoạt động đăng ký vắc xin – sinh phẩm y tế phải thoả mãn yêu cầu về tuyển dụng, đào tạo và theo dõi theo thủ tục của Cục về tuyển chọn nhân viên.
Cục Quản lý dược đánh giá nguồn nhân lực bao gồm cả đội ngũ chuyên gia thẩm định để đủ đáp ứng cho hoạt động của mình định kỳ qua đánh giá nội bộ và cuộc họp xem xét của lãnh đạo.
Mỗi cán bộ của Cục Quản lý dược tham gia vào hoạt động đăng ký vắc xin – sinh phẩm y tế có bản phân công nghiệp vụ cụ thể về nghĩa vụ, trách nhiệm và quyền hạn. Cán bộ và các chuyên gia thẩm định thực hiện công tác đăng ký vắc xin – sinh phẩm y tế phải thoả mãn yêu cầu về năng lực, trình độ và kinh nghiệm về quản lý cũng như về chuyên môn.
Cán bộ tham gia vào hoạt động đăng ký vắc xin – sinh phẩm y tế phải ký cam kết bảo mật theo quy định.
b) Nhân lực tham gia quá trình thẩm định
Chuyên gia thẩm định phải có chuyên môn, kinh nghiệm phù hợp cho lĩnh vực được phân công thẩm định.
Chuyên gia thẩm định phải được đào tạo về kỹ năng thẩm định hồ sơ đăng ký, có kiến thức về phương pháp thẩm định.
Chuyên gia thẩm định phải nắm được các chuẩn mực và quy định về thẩm định và phải có phẩm chất đạo đức nghề nghiệp thích hợp.
5.2- Đào tạo nhân lực:
Phòng Đăng ký thuốc có trách nhiệm tham mưu Lãnh đạo Cục về lập kế hoạch đào tạo cán bộ thực hiện công tác đăng ký vắc xin – sinh phẩm y tế, chuyên gia thẩm định với chương trình cụ thể hàng năm và tổ chức thực hiện khi kế hoạch, chương trình đã được Cục Trưởng phê duyệt.
Nội dung và hình thức đào tạo bao gồm (nhưng không giới hạn):
– Đào tạo nhằm duy trì và nâng cao trình độ chuyên môn, nghiệp vụ, kỹ năng thẩm định, khả năng ngoại ngữ phục vụ cho công tác, phục vụ nhu cầu tuyển chọn tiếp nhận nhân viên mới.
– Hình thức đào tạo có thể bao gồm đào tạo nội bộ, đào tạo bên ngoài. Tuỳ theo từng chuyên đề đào tạo có thời gian phù hợp với đối tượng và phải có chương trình cụ thể, rõ ràng.
Hàng năm việc xây dựng kế hoạch và đánh giá việc thực hiện kế hoạch đào tạo được xem xét trong đợt xem xét của lãnh đạo.
Kế hoạch đào tạo được lập theo biểu mẫu tại quy trình lựa chọn, sử dụng, đánh giá và đào tạo chuyên gia.
5.3. Theo dõi – đánh giá nguồn nhân lực
Cục Quản lý dược định kỳ tiến hành đánh giá toàn bộ nhân lực tham gia vào hoạt động đăng ký vắc xin – sinh phẩm y tế để đảm bảo hiệu quả thoả đáng của quá trình thẩm định, đánh giá (thông thường là 1 năm/lần đối với chuyên gia thẩm định, đánh giá), Cục Quản lý dược xem xét hoạt động và năng lực của cán bộ cũng như xác định nhu cầu đào tạo, cải tiến thích hợp, thay thế chuyên gia thẩm định khi không đảm bảo yêu cầu.
Mỗi lần dào tạo, phải có phiếu theo dõi đào tạo để lưu hồ sơ theo biểu mẫu tại quy trình lựa chọn, sử dụng, đánh giá và đào tạo chuyên gia
5.4. Hồ sơ nhân lực
Mỗi cán bộ tham gia thẩm định có hồ sơ thể hiện trình độ, quá trình công tác và quá trình đào tạo, kinh nghiệm và năng lực và hồ sơ thường xuyên được từng cán bộ cập nhật.
Cục Quản lý dược lập hồ sơ của toàn bộ chuyên gia thẩm định và chuyên gia kỹ thuật với đầy đủ nội dung theo biểu mẫu chung của Cục và cập nhật thông tin thường xuyên.
CHƯƠNG 5: QUÁ TRÌNH ĐĂNG KÝ VẮC XIN – SINH PHẨM Y TẾ
1- Sơ đồ đăng ký vắc xin – sinh phẩm y tế:
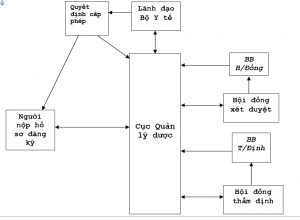
- Các quy định về đăng ký (Quy chế về đăng ký và các văn bản liên quan)
Cục Quản lý dược công khai quy định và các thông tin liên quan đến đăng ký vắc xin – sinh phẩm y tế cho các cá nhân, tổ chức liên quan biết và thực hiện (trên Web sites):
– Văn bản Quy phạm pháp luật;
– Thông tin về kết quả thẩm định;
– Các yêu cầu về hồ sơ;
– Phí đăng ký;
– Quyền lợi, trách nhiệm của các đơn vị được cấp phép và thông tin về các vắc xin – sinh phẩm y tế được cấp số đăng ký;
– Thông tin về khiếu nại và xem xét lại;
– Thông tin về các tổ chức có liên quan.
- Nộp đơn đề nghị cấp số đăng ký (Quy trình nộp hồ sơ đăng ký)
Đơn vị đăng ký nộp hồ sơ đăng ký lưu hành vắc xin – sinh phẩm y tế theo đúng quy định tại Văn phòng Cục.
- Xem xét hồ sơ
Cục Quản lý dược có trách nhiệm đảm bảo về nguồn lực cho hoạt động đăng ký thuốc theo các tiêu chí như chính sách, năng lực thực hiện, sự sẵn có của các chuyên gia đánh giá, đảm bảo kịp thời thời gian đánh giá.
- Nguyên tắc của hoạt động thẩm định, xét duyệt:
5.1. Tính công bằng
Cục Quản lý dược đảm bảo tính công bằng thông qua việc áp dụng một cơ cấu tổ chức thích hợp để đảm bảo cơ hội cho sự tham gia có hiệu lực của các bên quan tâm.
Toàn bộ chính sách và thủ tục về quá trình đăng ký vắc xin – sinh phẩm y tế được xây dựng đảm bảo không có sự phân biệt đối xử.
Tất cả nhân sự tham gia vào hoạt động đăng ký vắc xin – sinh phẩm y tế không chịu bất kỳ áp lực nào về kinh doanh, tài chính và các áp lực khác có thể ảnh hưởng đến tính công bằng.
Cục Quản lý dược đảm bảo các quyết định về đăng ký vắc xin – sinh phẩm y tế được đưa ra bởi những người có thẩm quyền, có năng lực thích hợp và không thực hiện trực tiếp hoạt động thẩm định.
Cục Quản lý dược không thực hiện bất kỳ dịch vụ tư vấn liên quan đến đăng ký vắc xin – sinh phẩm y tế.
Cục Quản lý dược đảm bảo hoạt động đăng ký vắc xin – sinh phẩm y tế không gây ảnh hưởng tới tính bảo mật, tính khách quan và tính công bằng của việc đăng ký.
Cục Quản lý dược thường xuyên báo cáo nêu rõ và phân tích các mối quan hệ với các tổ chức có liên quan để xác định khả năng xảy ra sự xung đột quyền lợi. Khi xung đột được nhận biết thì Cục Quản lý dược phải thực hiện hành động khắc phục thích hợp.
5.2. Tính bảo mật
Toàn bộ tổ chức, cá nhân tham gia hoạt động đăng ký vắc xin – sinh phẩm y tế phải thực hiện việc bảo mật theo quy định của pháp luật.
- Thực hiện quy trình đăng ký:
Thực hiện theo quy trình chung về đăng ký thuốc.
- Đánh giá bằng thực tế:
Yêu cầu đánh giá:
7.1- Sản xuất trong nước: Tất cả các nhà sản xuất vắc xin, sinh phẩm y tế trong nước đều được Cục Quản lý dược kiểm tra và cấp giấy chứng nhận GMP 2 năm 1 lần.
Ngoài ra có yêu cầu kiểm tra đột xuất, định kỳ khi có sự cố hoặc theo kế hoạch kiểm tra được xây dựng trên nguyên tắc quản lý rủi ro:
– Dây chuyền sản xuất:
+ Rủi ro cao: Vắc xin tiêm: Tần suất kiểm tra lớn hơn.
+ ít rủi ro hơn: Vắc xin uống: Tần suất kiểm tra thưa hơn.
– Theo quá trình sản xuất: Các công đoạn quan trọng thì tần suất kiểm tra lớn hơn so với các công đoạn sản xuất ít quan trọng hơn.
– Loại vắc xin: Vắc xin mới cần thường xuyên kiểm tra hơn so với vắc xin đã lưu hành lâu năm, ổn định chất lượng. Vắc xin sản xuất theo công nghệ an toàn hơn thì tuần suất kiểm tra ít hơn so với vắc xin có nhiều tác dụng phụ hơn… .
– Lượng sử dụng: Vắc xin có số lượng tiêu thụ, sử dụng nhiều thì cần thường xuyên kiểm tra hơn… .
Tần suất kiểm tra: bất cứ khi nào có sự cố; Hoặc định kỳ 01 lần/năm.
7.2- Sản xuất nước ngoài:
+ Chấp nhận theo quy định về công nhận lẫn nhau trong kiểm tra cấp giấy chứng nhận GMP trong khối ASEAN.
+ Danh sách các nước tham chiếu được chấp nhận: Anh, Pháp, Đức, Mỹ, Nhật Bản, úc, Canada hoặc EMEA (cơ quan thẩm định các sản phẩm y tế của Châu Âu).
+ Đối với các nước khác, trong trường hợp cần thiết Cục Quản lý Dược sẽ tiến hành đánh giá kiểm tra cơ sở sản xuất trước hoặc sau khi cấp số đăng ký lưu hành (Theo quy định tại Thông tư số 22 về việc đăng ký thuốc).
Đối với trường hợp cần đánh giá tại cơ sở sản xuất, Cục Quản lý dược có thể thành lập đoàn chuyên gia để tiến hành đánh giá.
Nhóm đánh giá phải thực hiện đánh giá theo chương trình chi tiết đã lập và được Cục Trưởng Cục Quản lý dược phê duyệt để đảm bảo tính pháp lý, khách quan.
- Các yêu cầu xem xét lại các khiếu nại
Bất kỳ một yêu cầu xem xét lại của các đơn vị liên quan đến kết luận của quá trình thẩm định, xét duyệt và các quyết định của Cục Quản lý dược phải được giải quyết. Khi có vấn đề cần xem xét lại, các đơn vị có công văn đề nghị Cục Quản lý dược xem xét lại.
Khi có yêu cầu về xem xét lại của các công ty, Cục Trưởng Cục Quản lý dược sẽ đề nghị các cá nhân, tổ chức liên quan tiến hành việc xem xét các yêu cầu này của đơn vị.
Cán bộ được phân công xem xét giải quyết khiếu nại phải đảm bảo có năng lực và độc lập với chủ thể yêu cầu xem xét lại.
Việc xem xét lại phải đưa ra được kết luận và quyết định về tính hiệu lực, phải thông báo cho các bên có liên quan về quyết định cuối cùng liên quan đến vấn đề yêu cầu xem xét lại.
Hồ sơ về các yêu cầu xem xét lại, quyết định cuối cùng, kiểm tra (nếu có) được lưu giữ tại Cục Quản lý dược.
Sai sót được rà soát theo biểu mẫu 01 kèm theo Sổ tay này.
Thời gian xem xét: trong vòng 20 ngày làm việc. Nếu lâu hơn phải nêu rõ lý do.
9- Thực hiện quy trình đăng ký nhanh:
9.1- Tiêu chí:
– Vắc xin, sinh phẩm y tế cần thiết dùng trong đại dịch.
– Các vắc xin, sinh phẩm y tế có nhu cầu sử dụng khẩn cấp.
9.2- Thực hiện quy trình: Thực hiện theo các nguyên tắc sau:
– Nội dung hồ sơ: Chuẩn bị theo quy định hiện hành. Tuy nhiên để rút ngắn được thời gian đăng ký có thể chấp nhận tối thiểu như sau:
+ Các đề mục hồ sơ phải đủ như quy định. Nội dung của từng phần có thể mới có các nội dung cơ bản, quan trọng nhưng chưa hoàn toàn đầy đủ.
+ Có thể chấp nhận áp dụng các Format của ACTD, ICH-CTD.
+ Trong thời gian thẩm định hồ sơ, công ty có thể bổ sung hồ sơ đầy đủ mà không đợi thông báo kết quả thẩm định.
+ Cuối cùng công ty phải bổ sung đầy đủ hồ sơ theo qui định.
– Thẩm định hồ sơ:
+ Thời gian thẩm định: ưu tiên thẩm định, không phải đợi đến thứ tự thẩm định. Thời gian thẩm định nhanh nhất có thể.
+ Thẩm định trên nội dung tài liệu do công ty nộp ban đầu và nộp bổ sung dần, bổ sung hoàn chỉnh.
– Thông báo kết quả thẩm định: Khi từng nhóm thẩm định xong, không nhất thiết phải thẩm định xong tất cả các nhóm. Hình thức thông báo có thể họp trực tiếp hoặc bằng văn bản nhưng thông báo cuối cùng phải bằng văn bản.
– Yêu cầu về lâm sàng tại Việt Nam: Cục Quản lý Dược có văn bản chuyển hồ sơ liên quan để Vụ Khoa học và Đào tạo xin ý kiến các Hội đồng liên quan đến thử lâm sàng để trình Bộ Trưởng Quyết định chính thức về miễn thử hoặc miễn một số giai đoạn.
– Hồ sơ thẩm định xong sẽ được trình Hội đồng xét duyệt và báo cáo Lãnh đạo Bộ xem xét theo quy định.
CHƯƠNG 6: ĐẢM BẢO CHẤT LƯỢNG TRONG ĐĂNG KÝ VẮC XIN – SINH PHẨM Y TẾ
- Quy định chung:
Cục Quản lý dược luôn duy trì áp dụng hệ thống quản lý và cải tiến liên tục hiệu lực của hệ thống phù hợp tiêu chuẩn ISO và hoà hợp quốc tế.
Cục Quản lý dược ban hành và áp dụng tài liệu hệ thống quản lýý bằng văn bản bao gồm: sổ tay chất lượng, các thủ tục, các quy định, các hướng dẫn phù hợp yêu cầu của ISO.
- Hệ thống quản lý
Hệ thống quản lý của Cục Quản lý dược hoàn toàn tuân thủ tiêu chuẩn ISO và các quy định có liên quan khác.
Chính sách chất lượng và mục tiêu chung được lãnh đạo Cục Quản lý dược công bố trong sổ tay chất lượng. Mục tiêu chất lượng hàng năm được đề xuất trong cuộc họp của lãnh đạo Cục.
Hệ thống quản lý của Cục Quản lý dược được lập thành văn bản, được phổ biến và đảm bảo việc áp dụng của các cá nhân có liên quan đến đăng ký vắc xin – sinh phẩm y tế. Hệ thống tài liệu của Cục Quản lý dược gồm: Sổ tay chất lượng, các thủ tục, các hướng dẫn, các biểu mẫu, các hồ sơ và báo cáo liên quan đến đăng ký vắc xin – sinh phẩm y tế. Ngoài ra Cục Quản lý dược còn lưu giữ và áp dụng các tài liệu khác bao gồm:
– Tiêu chuẩn ISO về quản lý hành chính;
– Các văn bản pháp quy kỹ thuật của Nhà nước liên quan đến hoạt động quản lý thuốc, vắc xin – sinh phẩm y tế;
– Các tài liệu tham khảo về hoà hợp quy chế trong khu vực.
Cục Quản lý dược đảm bảo hoạt động của hệ thống quản lýý và luôn cải tiến thông qua các hoạt động như: họp định kỳ, đánh giá nội bô, xem xét của lãnh đạo, hành động khắc phục, phòng ngừa, tiếp nhận và giải quyết khiếu nại …
- Kiểm soát tài liệu
Cục Quản lý dược thực hiện kiểm soát tất cả các tài liệu nội bộ và bên ngoài liên quan đến hoạt động đăng ký vắc xin – sinh phẩm y tế. Thủ tục này đảm bảo tài liệu được soát xét, cập nhật, phê duyệt trước khi ban hành, tài liệu được phổ biến kịp thời, đúng đối tượng và các yêu cầu liên quan kiểm soát tài liệu theo quy định của ISO.
- Sự không phù hợp và hành động khắc phục.
Cục Quản lý dược thực hiện việc giám sát, phát hiện các điểm không phù hợp, thực hiện hành động khắc phục đảm bảo: Các hành động thích hợp để loại bỏ các nguyên nhân gây ra sự không phù hợp nhằm ngăn ngừa sự tái diễn. Rà soát theo biểu mẫu 01 kèm theo Sổ tay này
- Hành động phòng ngừa
Cục Quản lý dược thực hiện các biện pháp phòng ngừa sau:
– Phát hiện các điểm không phù hợp tiềm ẩn và nguyên nhân gây ra chúng.
– Đưa ra các biện pháp phòng ngừa cần thiết.
– Lập hồ sơ kết quả của các hành động đã thực hiện;
– Xem xét kết quả của các hành động phòng ngừa đã thực hiện.
- Đánh giá nội bộ
Đánh giá nội bộ thực hiện định kỳ một năm/ lần, nội dung đánh giá bao gồm toàn bộ các yếu tố của hệ thống quản lý để đảm bảo hệ thống quản lýý được duy trì và cải tiến.
Chuyên gia đánh giá nội bộ là người có trình độ và am hiểu về công nhận, đánh giá và các yêu cầu của ISO và khách quan với hoạt động được đánh giá.
Biện pháp khắc phục (nếu có điểm không phù hợp) phải được thực hiện kịp thời và thông qua đánh giá nội bộ, qua đó có thể cải tiến.
- Xem xét của lãnh đạo
Xem xét của lãnh đạo thực hiện định kỳ một năm/ lần, đầu vào của xem xét của lãnh đạo bao gồm hoạt động hiện thời và biện pháp cải tiến liên quan đến đăng ký vắc xin – sinh phẩm y tế.
Đầu ra của xem xét của lãnh đạo phải bao gồm: cải tiến hệ thống quản lý đăng ký vắc xin – sinh phẩm y tế của Cục Quản lý dược, xác định nhu cầu về nguồn lực và xác định lại chính sách và mục tiêu chất lượng.
- ý kiến phản hồi
Cục Quản lý dược thực hiện tiếp nhận và giải quyết các ý kiến phản hồi đảm bảo ý kiến liên quan hoạt động đăng ký vắc xin – sinh phẩm y tế sẽ được Cục Quản lý dược thông báo tới cá nhân, tổ chức liên quan để xử lý trước và xử lý một cách phù hợp, kịp thời.
PHỤ LỤC 1
HƯỚNG DẪN THẨM ĐỊNH HỒ SƠ ĐĂNG KÝ LƯU HÀNH VẮC XIN – SINH PHẨM Y TẾ
1- Mục đích:
Hồ sơ đăng ký vắc xin và sinh phẩm y tế phải được thẩm định trước khi trình Hội đồng xét duyệt hồ sơ đăng ký lưu hành tại Việt Nam. Hướng dẫn bao gồm quy trình cụ thể nhằm đảm bảo hồ sơ được xem xét một cách khoa học, trung thực, khách quan.
2- Các nhóm chuyên gia thẩm định: Theo Quyết định của Cục Trưởng Cục Quản lý Dược.
Bao gồm các chuyên gia thẩm định gồm các lĩnh vực:
– Chuyên gia về hồ sơ hành chính.
– Chuyên gia về hồ sơ chất lượng.
– Chuyên gia về hồ sơ an toàn và hiệu quả.
Trưởng các nhóm chuyên gia do Cục Quản lý dược chỉ định.
3- Thẩm định của chuyên gia
B1- Xác định phần hồ sơ thẩm định:
Chuyên gia thuộc từng lĩnh vực thẩm định xác định trong hồ sơ đăng ký những phần tài liệu thuộc nhóm của mình thẩm định.
B2- Thẩm định hồ sơ: Lần đầu và thẩm định bổ sung.
– Căn cứ để thẩm định: Các quy chế, quy định liên quan đến đăng ký vắc xin; Tài liệu hướng dẫn đăng ký vắc xin; Các dược điển; Các hướng dẫn, tài liệu chuyên môn của WHO; Các tài liệu chuyên môn về Y – dược – sinh học; Trình độ, kinh nghiệm của chuyên gia.
– Tính đầy đủ của hồ sơ: Kiểm tra và xác định sự đầy đủ của hồ sơ thuộc lĩnh vực thẩm định theo quy định về đăng ký vắc xin hiện hành. Xác định trạng thái của các phần trong hồ sơ: có hoặc không.
– Tính đúng và chính xác của hồ sơ: Các vấn đề nêu ra trong hồ sơ phải có bằng chứng rõ ràng; Hồ sơ phải đảm bảo chính xác, khoa học, trung thực, khách quan.
– Tính áp dụng vào thực tế: Các phần hồ sơ phải áp dụng được vào thực tế quản lý cũng như trong chuyên môn, kỹ thuật và sử dụng.
– Thống nhất ý kiến thẩm định: các thành viên trong nhóm thảo luận ý kiến nhận xét của từng thành viên và Trưởng nhóm kết luận.
– Ghi ý kiến vào biên bản thẩm định sau khi đã thảo luận trong nhóm.
Sơ đồ liên kết giữa các tiểu ban
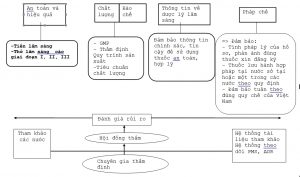
4- Yêu cầu cụ thể thẩm định hồ sơ đăng ký vắc xin các tiểu ban kỹ thuật
4.1. Phần hồ sơ về chất lượng:
Cần thẩm định các vấn đề chủ yếu sau:
| Đề mục | Yêu cầu | Hướng dẫn thêm |
| 2.1- Mục lục của phần hồ sơ chất lượng | ||
| 2.2- Nội dung: | ||
| 2.2.1- Thành phần hoạt chất: | – Cung cấp thông tin cho mỗi thành phần hoạt chất trong vắc xin
– Hoạt chất của vắc xin: Các chất kháng nguyên (hoặc hợp chất của nó) gây ra các đáp ứng đặc hiệu trong cơ thể người nhằm chống lại các tác nhân truyền nhiễm, các kháng nguyên hoặc các độc tính của nó |
|
| 2.2.1.1- Thông tin chung, nguyên liệu đầu vào và nguyên liệu thô
– Nguyên liệu thô: bất kỳ chất nào được sử dụng để tạo ra hoặc chiết ra thành phần hoạt chất nhưng thành phần hoạt chất không phải là dẫn xuất trực tiếp từ đó. Ví dụ: môi trường nuôi cấy, hyuết thanh súc vật… . – Nguyên liệu đầu vào: bất kỳ chất nào có nguồn gốc sinh học, như vi sinh vật, các tổ chức, mô có nguồn gốc động vật, thực vật, bao gồm tế bào, dịch có nguồn gốc từ người, hoặc động vật và các tế bào tái tổ hợp. |
– Tên thương mại và/hoặc tên chung của thành phần hoạt chất.
– Công thức cấu tạo, công thức phân tử và trọng lượng phân tử liên quan (nếu có). – Mô tả và đặc trưng của thành phần hoạt chất. – Phiếu kiểm nghiệm. – Mô tả chung của nguyên liệu đầu vào: Chủng; Hệ thống chủng/ngân hàng chủng gốc/ngân hàng chủng sản xuất; Trứng gà kết phôi. – Mô tả chung của nguyên liệu thô. |
– Đặc trưng lý hoá: Những đặc trưng lý hoá chủ chốt (ví dụ: hàm lượng nước, tính hoà tan, kích thước tiêu phân, đa hình thái, trạng thái hydrat hoá, pH, hằng số điện ly (pKa) của hoạt chất |
| 2.2.1.2- Qui trình sản xuất của thành phần hoạt chất: | – Các nhà sản xuất liên quan.
– Mô tả quy trình sản xuất. – Sơ đồ quy trình sản xuất. – Mô tả hệ thống nhận dạng lô. – Xác định những bước quan trọng trong quy trình và kiểm soát. – Mô tả quy trình bất hoạt và giải độc tố – Mô tả quy trình tinh chế – Mô tả quy trình kết hợp – làm ổn định thành phần hoạt chất. – Chế biến lại – Quy trình đóng (ống, lọ…) đối với hoạt chất, kiểm soát trong quá trình sản xuất. – Lựa chọn và chứng minh các bước quan trọng. – Thẩm định quy trình sản xuất – Mô tả sự thay đổi |
– Nhà SX: Tên và địa chỉ đầy đủ bao gồm thành phố, tên nước của nhà sản xuất.
– Mô tả Quy trình SX: Thông tin về quá trình sản xuất bắt đầu bằng ngân hàng tế bào và bao gồm nuôi cấy tế bào, gặt, tinh chế -Nguyên liệu sử dụng trong sản xuất (VD: nguyên liệu thô, nguyên liệu đầu vào, dung môi, thuốc thử, chất xúc tác) phải được liệt kê và xác định rõ được sử dụng ở đâu trong quy trình sản xuất. Thông tin về chất lượng và kiểm soát chất lượng của những nguyên liệu này. Thông tin chứng minh các nguyên liệu (bao gồm những nguyên liệu nguồn gốc sinh học,… các thành phần môi trường, các kháng thể đơn dòng, các men) đạt tiêu chuẩn phù hợp với mục đích sử dụng. |
| 2.2.1.3- Kiểm tra chất lượng của thành phần hoạt chất | – Mô tả quy trình phân tích, thẩm định và chứng minh tiêu chuẩn. | – Tiêu chuẩn: Đặc điểm kỹ thuật chi tiết, các phép thử và các chỉ tiêu đối với thành phần hoạt chất.
+ Chỉ rõ nguồn như chủng, loài động vật, loại vi sinh… . – Quy trình phân tích: Cần phải mô tả quy trình chi tiết để các phòng kiểm nghiệm theo đó có thể tiến hành được. – Thẩm định phương pháp phân tích: bao gồm các dữ liệu thử nghiệm cho quy trình phân tích. Thẩm định phương pháp phân tích xem xét các khía cạnh như độ chọn lọc, độ đúng, độ chính xác, độ tuyến tính, giới hạn số lượng, giới hạn phát hiện, sự phù hợp của hệ thống… . |
| 2.2.1.4- Độ ổn định của thành phần hoạt chất | – Đề cương nghiên cứu, kết quả nghiên cứu và kết luận
– Điều kiện bảo quản
|
– Mô tả tóm tắt loại nghiên cứu, đề cương và kết quả của các nghiên cứu. Kết luận tuổi thọ phải phù hợp với điều kiện bảo quản.
– Kết quả nghiên cứu cần phải được thể hiện bằng bản biểu, đồ thị… . |
| 2.2.1.5- Sự ổn định của sản xuất thành phần hoạt chất. | ||
| 2.2.2- Thành phẩm | ||
| 2.2.2.1- Mô tả và công thức của thành phẩm | Bao gồm những thông tin:
– Mô tả dạng bào chế; – Thành phần: danh mục tất cả các thành phần của dạng bào chế và số lượng của chúng trong 1 đơn vị (bao gồm cả số dư, nếu có), chức năng của các thành phần, tiêu chuẩn tham chiếu. – Mô tả dung môi pha loãng kèm theo; – Loại bao bì đóng gói và nắp đậy. |
|
| 2.2.2.2- Sự phát triển sản phẩm | – Thành phần hoạt chất
– Thành phẩm – Quy trình sản xuất – Tính tương thích, hệ thống nắp đậy/bao gói
|
– Sự phát triển sản phẩm bao gồm thông tin về những nghiên cứu được tiến hành để thiết lập nên dạng bào chế, công thức, qui trình sản xuất, hệ thống nắp đậy, bao gói, các đặc tính vi sinh, hướng dẫn sử dụng.
– |
| 2.2.2.3- Sản xuất thành phẩm | – Nhà sản xuất
– Công thức lô – Mô tả quy trình sản xuất – Kiểm soát các bước quan trọng và các bước trung gian – Thẩm định và đánh giá quy trình – Mô tả hệ thống nhận dạng lô |
– Nhà SX: Tên và địa chỉ đầy đủ bao gồm thành phố, tên nước của nhà sản xuất.
– Công thức lô: Bao gồm tên và số lượng của tất cả thành phần (hoạt chất và tá dược): Số lượng thực tế (g, kg…); Dư thừa: cung cấp số liệu và lý do của sự dư thừa; Số đơn vị liều trong 1 lô. – Quy trình sản xuất: Sơ đồ các bước sản xuất (phải chỉ rõ nơi nguyên liệu được đưa vào quy trình); Các bước quan trọng và các điểm phải kiểm soát; xác định các phép thử trung gian hoặc sản phẩm cuối cùng. Mô tả chi tiết tất cả các bước sản xuất (bao gồm các điểm thiết yếu của mỗi giai đoạn). Đối với sản phẩm vô khuẩn mô tả phải bao gồm sự chuẩn bị và vô khuẩn của các thành phần (bao bì, nắp… ). – Các bước quan trọng: Cung cấp các phép thử và các chỉ tiêu (có tài liệu chứng minh, dữ liệu thử nghiệm) được tiến hành tại các bước quan trọng của quy trình sản xuất nhằm bảo đảm quy trình sản xuất được kiểm soát. Trung gian: Thông tin về chất lượng và kiểm soát của sản phẩm trung gian được biệt lập trong quá trình sản xuất. – Thẩm định và đánh giá quy trình: Mô tả, cung cấp tài liệu, và kết quả của các nghiên cứu đánh giá tại các bước quan trọng hoặc các phân tích quan trọng trong quá trình sản xuất. |
| 2.2.2.4- Kiểm soát tá dược, chất bảo quản, chất ổn định | – Tiêu chuẩn
– Quy trình phân tích – Thẩm định quy trình phân tích – Chứng minh tiêu chuẩn – Các chất có nguồn gốc từ người, động vật – Sử dụng tá dược, chất bảo quản, chất ổn định mới |
– Nguyên liệu có nguồn gốc từ người và động vật: bao gồm nguồn, tiêu chuẩn, mô tả phương pháp thử, dữ liệu an toàn vi rút.
– Sử dụng tá dược mới: phải cung cấp đầy đủ chi tiết về sản xuất, đặc tính và kiểm soát, với sự tài liệu tham khảo về an toàn (tiền lâm sàng, lâm sàng). |
| 2.2.2.5- Kiểm soát thành phẩm | 2.2.2.5.1- Tiêu chuẩn
2.2.2.5.2- Quy trình phân tích 2.2.2.5.3- Phiếu kiểm nghiệm của nhà sản xuất 2.2.2.5.4- Thẩm định quy trình phân tích 2.2.2.5.5- Tính ổn định và sự phân tích lô 2.2.2.5.6- Xác định và đặc trưng của tạp chất 2.2.2.5.7- Chứng minh tiêu chuẩn |
– Tiêu chuẩn: Cung cấp tiêu chuẩn, chỉ tiêu của thành phẩm.
– Quy trình phân tích: Quy trình phân tích của từng phép thử trong tiêu chuẩn thành phẩm. – Thẩm định quy trình phân tích: Thông tin thẩm định phân tích bao gồm dữ liệu thử nghiệm cho quy trình phân tích của các phép thử. – Tính ổn định và sự phân tích lô: Mô tả (bao gồm cỡ lô, nguồn gốc và sử dụng) và kết quả thử nghiệm của tất cả các lô thích hợp (VD:) sử dụng để thiết lập tiêu chuẩn và đánh giá sự ổn định trong sản xuất |
| 2.2.2.6- Tiêu chuẩn hoặc nguyên liệu tham chiếu | – Thông tin chất lượng và liệt kê các tiêu chuẩn tham chiếu hoặc nguyên liệu sử dụng cho các phép thử. | |
| 2.2.2.7- Hệ thống nắp đậy/ bao gói | – Tiêu chuẩn của bao bì trực tiếp và gián tiếp
– Thử nghiệm và đánh giá nguyên liệu bao bì |
– Mô tả hệ thống bao bì, nắp đậy bao gồm: xác định nguyên liệu bao bì trực tiếp, bao bì gián tiếp, tiêu chuẩn. Tiêu chuẩn bao gồm mô tả và định tính, kích thước, phương pháp kiểm tra. |
| 2.2.3- Độ ổn định | ||
| 2.2.3. 1- Đề cương của nghiên cứu, kết quả và kết luận | – Đối với sản phẩm đông khô, bao gồm nghiên cứu độ ổn định của nguyên liệu đông khô, dung môi và sản phẩm hoàn nguyên
– Tính chịu nhiệt (nếu có) |
– Mô tả tóm tắt loại nghiên cứu, đề cương và kết quả của các nghiên cứu. Kết luận tuổi thọ phải phù hợp với điều kiện bảo quản.
– Kết quả nghiên cứu cần phải được thể hiện bằng bảng biểu, đồ thị… . |
| 2.2.3.2- Mô tả quy trình bảo đảm dây chuyền lạnh | ||
| 3.2.4- Danh mục trang thiết bị sản xuất, kiểm định |
4.2. Phần hồ sơ về an toàn (tiền lâm sàng): cần xem xét các nội dung chủ yếu sau:
| Đề mục | Yêu cầu | Hướng dẫn thêm |
| 3.1- Mục lục của phần hồ sơ an toàn | ||
| 3.2- Tóm tắt về tiền lâm sàng | – Giới thiệu
– Viết tóm tắt về dược lý – Bảng tóm tắt về dược lý – Viết tóm tắt về dược động học (nếu có) – Bảng tóm tắt về dược động học (nếu có) – Viết tóm tắt về độc tính – Bảng tóm tắt về độc tính.
|
|
| 3.3- Các báo cáo nghiên cứu | ||
| 3.3.1- Dược lý | 3.2.1.1- Nghiên cứu dược lực học (Kháng nguyên của vắc xin)
3.2.1.2- Dược lực học của tá dược (adjuvant) – nếu có |
|
| 3.3.2- Dược động học | 3.2.2.1- Các nghiên cứu dược động học (trong trường hợp các tá dược mới, đường dùng mới) | |
| 3.3.3- Độc tính | ||
| 3.3.3.1- Độc tính chung | – Thiết kế nghiên cứu và chứng minh mẫu động vật
– Loài động vật sử dụng, tuổi, kích cỡ của nhóm – Liều, đường dùng và kiểm soát nhóm – Các thông số theo dõi – Tính dung nạp tại chỗ |
|
| 3.3.3.2- Độc tính đặc biệt | – Những nghiên cứu miễn dịch đặc biệt
– Những nghiên cứu độc tính trên những dân số đặc biệt – Những nghiên cứu độc tính gen và gây ung thư |
|
| 3.3.3.3- Độc tính của các chất mới sử dụng trong công thức (các tá dược mới, chất ổn định, chất độn) | ||
| 3.3.4- Những xem xét đặc biệt | 3.2.4.1- Đối với các vắc xin pha loãng, đánh giá khả năng “rơi ra” (chất bài tiết) của vi sinh vật
3.2.4.2- Độc tính của các chất mới sử dụng trong công thức (tá dược mới, chất ổn định mới, chất độn mới), đường dùng khác hoặc các vắc xin kết hợp – cung cấp nhứng nghiên cứu độc tính thích hợp |
4.3. Phần hồ sơ về hiệu quả (lâm sàng):
Cần xem xét những nội dung chủ yếu sau:
| Đề mục | Yêu cầu | Hướng dẫn thêm |
| 4.1- Mục lục của phần hồ sơ lâm sàng | ||
| 4.2- Tổng quan về nghiên cứu lâm sàng | – Giới thiệu
– Mục lục – Tổng quan về kháng nguyên – Tổng quan về hiệu quả – Tổng quan về an toàn – Kết luận về cân bằng lợi ích – rủi ro – Thư mục tham khảo |
|
| 4.3- Tóm tắt về lâm sàng | – Giới thiệu
– Mục lục – Tóm tắt những nghiên cứu lâm sàng của kháng nguyên – Tóm tắt các nghiên cứu lâm sàng về hiệu quả – Tóm tắt những nghiên cứu lâm sàng về an toàn – Thư mục tham khảo |
|
| 4.3- Các báo cáo nghiên cứu | ||
| 4.3.1- Nghiên cứu giai đoan I | ||
| 4.3.2- Nghiên cứu giai đoan II | ||
| 4.3.3- Nghiên cứu giai đoan III | ||
| 4.3.4- Những xem xét đặc biệt | ||
| 4.3.5- Các tá dược | ||
| 4.3.6- Nghiên cứu giai đoạn IV và / hoặc kế hoạch dược cảnh giác (nếu có) | ||
| 4.3.7- Những nghiên cứu về không giảm hiệu quả đối với các vắc xin kết hợp, hoặc các vắc xin được sản xuất ở nhà sản xuất mới. | ||
| 4.3.8- Những nghiên cứu về sử dụng đồng thời với vắc xin khác |
Biểu mẫu 1: Kế hoạch rà soát quy trình để tìm ra các sai sót
| Các hoạt động rà soát liên quan đến qui trình đăng ký vắc xin – sinh phảm y tế | Đạt theo quy định | Không đạt theo qui định | Hành động khắc phục | Ghi chú |
| 1- Hoạt động 1: Tiếp nhận hồ sơ và thu phí đăng ký:
– Rà soát từng bước theo theo quy trình tiếp nhận hồ sơ Đăng ký thuốc (Quy trình số QT.QLD.11)
|
||||
| 2- Hoạt động 2: Phân loại và chuẩn bị hồ sơ đưa ra thẩm định:
– Rà soát từng bước theo Quy trình chuẩn bị hồ sơ (số QT.QLD.12).
|
||||
| 3- Hoạt động 3: Thẩm định hồ sơ đăng ký thuốc: Rà soát theo các nội dung thẩm định sau:
– Thuốc mới, thuốc từ dược liệu mới và vắc xin – sinh phẩm y tế: Thẩm định các phần hồ sơ Hành chính – Thông tin sản phẩm; Hồ sơ chất lượng; Hồ sơ an toàn và hiệu quả – Thông tin liên quan đến sử dụng thuốc hợp lý, an toàn. – Thuốc generic, thuốc đông y: Thẩm định các phần hồ sơ Hành chính – Thông tin sản phẩm; Hồ sơ chất lượng; Thông tin liên quan đến sử dụng thuốc hợp lý, an toàn.
|
||||
| 4- Hoạt động 4: Xử lý hồ sơ sau thẩm định:
– Rà soát các bước theo Quy trình xử lý hồ sơ ĐKT sau khi thẩm định (QT.QLD.13);
|
||||
| 5- Hoạt động 5: Chuẩn bị nội dung và xử lý biên bản họp Hội đồng:
– Rà soát các bước theo Quy trình xử lý biên bản họp HĐXDT (QT. QLD.15);
|
||||
| 6- Hoạt động 6: Chuẩn bị Danh mục và ban hành Quyết định cấp SĐK:
– Rà soát từng bước theo Quy trình chuẩn bị và ký Quyết định cấp SĐK. (QT.QLD 16);
|
Sổ tay chất lượng đăng ký vắc xin sinh phẩm y tế
DOWNLOAD FILE DƯỚI ĐÂY
[sociallocker id=6157]
FILE WORD ĐÍNH KÈM: SO TAY CHAT LUONG VACCIN BAN BAN HANH
[/sociallocker]












{kind=link}