









")













Quyết định 2111/QĐ-BYT ban hành hướng dẫn quốc gia về cảnh giác Dược
| BỘ Y TẾ ——– |
CỘNG HÒA XÃ HỘI CHỦ NGHĨA VIỆT NAM Độc lập – Tự do – Hạnh phúc ————— |
| Số: 2111/QĐ-BYT | Hà Nội, ngày 01 tháng 6 năm 2015 |
QUYẾT ĐỊNH
VỀ VIỆC BAN HÀNH HƯỚNG DẪN QUỐC GIA VỀ CẢNH GIÁC DƯỢC
BỘ TRƯỞNG BỘ Y TẾ
Căn cứ Nghị định số 63/2012/NĐ-CP ngày 31/8/2012 của Chính phủ quy định chức năng, nhiệm vụ, quyền hạn và cơ cấu tổ chức của Bộ Y tế;
Xét đề nghị của Cục trưởng Cục Quản lý Dược – Bộ Y tế,
QUYẾT ĐỊNH:
Điều 1. Ban hành kèm theo Quyết định này “Hướng dẫn Quốc gia về Cảnh giác Dược”.
Điều 2. Quyết định này có hiệu lực kể từ ngày ký ban hành.
Điều 3. Các Ông, Bà: Chánh Văn phòng Bộ; Chánh Thanh tra Bộ; Cục trưởng Cục Quản lý Dược; Vụ trưởng, Cục trưởng, Tổng Cục trưởng các Vụ, Cục, Tổng cục thuộc Bộ Y tế; Thủ trưởng các đơn vị có liên quan chịu trách nhiệm thi hành Quyết định này./.
|
Nơi nhận: |
KT. BỘ TRƯỞNG THỨ TRƯỞNG Lê Quang Cường |
BỘ Y TẾ
HƯỚNG DẪN QUỐC GIA VỀ CẢNH GIÁC DƯỢC
Hà Nội – 2015
BAN BIÊN SOẠN
(Được thành lập theo Quyết định số 3551/QĐ-BYT, ngày 19/09/2013 của Bộ trưởng Bộ Y tế)
- Ông Lê Quang Cường, Thứ trưởng Bộ Y tế: Trưởng ban
- Ông Trương Quốc Cường, Cục trưởng Cục Quản lý Dược, Bộ Y tế: Phó trưởng ban
- Ông Nguyễn Huy Quang, Vụ trưởng Vụ Pháp chế, Bộ Y tế
- Ông Cao Hưng Thái, Phó Cục trưởng Cục Quản lý Khám, chữa bệnh, Bộ Y tế
- Bà Nguyễn Minh Hằng, Phó Cục trưởng Cục Y tế dự phòng, Bộ Y tế
- Ông Nguyễn Ngô Quang, Phó Cục trưởng Cục Khoa học công nghệ & Đào tạo, Bộ Y tế
- Bà Trần Thị Hồng Phương, Phó Cục trưởng Cục Quản lý Y, Dược cổ truyền, Bộ Y tế
- Ông Bùi Đức Dương, Phó Cục trưởng Cục Phòng, chống HIV/AIDS, Bộ Y tế
- Ông Nguyễn Đăng Hòa, Hiệu trưởng Trường Đại học Dược Hà Nội, Giám đốc Trung tâm Quốc gia về thông tin thuốc và theo dõi phản ứng có hại của thuốc
- Ông Nguyễn Hoàng Anh, Phó Giám đốc Trung tâm Quốc gia về thông tin thuốc và theo dõi phản ứng có hại của thuốc, Trường Đại học Dược Hà Nội
- Ông Trần Thanh Dương, Viện trưởng Viện Sốt rét – Ký sinh trùng – Côn trùng Trung ương
- Ông Trần Việt Hùng, Phó Viện trưởng Viện Kiểm nghiệm thuốc Trung ương
- Bà Trương Thị Thu Lan, Phó Viện trưởng Viện Kiểm nghiệm thuốc thành phố Hồ Chí Minh
- Ông Nguyễn Quốc Bình, Phó Giám đốc Trung tâm khu vực về thông tin thuốc và theo dõi phản ứng có hại của thuốc thành phố Hồ Chí Minh, Bệnh viện Chợ Rẫy
- Ông Vũ Xuân Phú, Phó Giám đốc Bệnh viện Phổi Trung ương
- Bà Phạm Thị Thúy Vân, Phó trưởng Bộ môn Dược lâm sàng, Trường Đại học Dược Hà Nội
- Bà Trương Thị Nguyệt, Phó Trưởng phòng Quản lý Thông tin quảng cáo thuốc, Cục Quản lý Dược, Bộ Y tế
Tổ thư ký
1. Ông Bùi Quang Phúc, Trưởng khoa Nghiên cứu điều trị sốt rét, Viện Sốt rét – Ký sinh trùng – Côn trùng Trung ương
- Bà Ngô Thị Bích Hà, Phó trưởng phòng Nghiệp vụ – Pháp chế, Cục Quản lý Khám, chữa bệnh, Bộ Y tế
- Ông Nguyễn Xuân Tùng, Phó trưởng phòng Vắc xin, sinh phẩm y tế và an toàn sinh học, Cục Y tế dự phòng, Bộ Y tế
- Bà Lê Thị Luyến, Chuyên viên chính, Cục Khoa học công nghệ & Đào tạo, Bộ Y tế
- Bà Cao Thị Mai Phương, Nguyên Giám đốc Trung tâm Dược điển Dược thư Việt Nam, Bộ Y tế
- Bà Lê Thị Hường, Phó Trưởng phòng Điều trị và chăm sóc HIV/AIDS, Cục Phòng chống HIV/AIDS, Bộ Y tế
- Bà Hoàng Thanh Mai, Phó trưởng phòng Quản lý Thông tin quảng cáo thuốc, Cục Quản lý Dược, Bộ Y tế
- Bà Nguyễn Thị Thủy, Phó trưởng khoa Dược, Bệnh viện Phổi Trung ương
- Bà Cao Thị Cẩm Tú, Kiểm nghiệm viên, Trung tâm đánh giá Tương đương sinh học, Viện Kiểm nghiệm thuốc thành phố Hồ Chí Minh
- Bà Nguyễn Thị Phương Lan, Phó trưởng phòng Quản lý Dược cổ truyền, Cục Quản lý Y, Dược cổ truyền, Bộ Y tế
- Bà Châu Ánh Minh, Cán bộ Trung tâm khu vực về thông tin thuốc và theo dõi phản ứng có hại của thuốc thành phố Hồ Chí Minh, Bệnh viện Chợ Rẫy
- Bà Nguyễn Thị Minh Thu, Nghiên cứu viên khoa Nghiên cứu điều trị sốt rét, Viện Sốt rét – Ký sinh trùng – Côn trùng Trung ương
- Bà Võ Thị Thu Thủy, Phó Giám đốc Trung tâm Quốc gia về thông tin thuốc và theo dõi phản ứng có hại của thuốc, Trường Đại học Dược Hà Nội
LỜI NÓI ĐẦU
Sự ra đời của nhiều thuốc mới đã mang lại lợi ích to lớn trong điều trị và chăm sóc sức khỏe cho cộng đồng, song cũng đặt ra nhiều thách thức trong công tác đảm bảo sử dụng thuốc hợp lý, an toàn và hiệu quả. Trong những năm gần đây, nhiều thuốc đã bị rút khỏi thị trường dược phẩm do khi sử dụng nguy cơ cao hơn hẳn lợi ích mà thuốc mang lại. Với mục đích giảm thiểu tác động có hại của thuốc đối với cộng đồng, hệ thống Cảnh giác dược đã được hình thành và phát triển rộng khắp tại nhiều quốc gia nhằm phát hiện, theo dõi, đánh giá và phòng tránh những biến cố bất lợi cũng như các vấn đề khác liên quan đến sử dụng thuốc. Tại Việt Nam, từ năm 1994, hệ thống Cảnh giác dược đã bước đầu hình thành bằng việc thiết lập mạng lưới thông tin thuốc và theo dõi phản ứng có hại của thuốc. Đến nay, hoạt động Cảnh giác dược đã được quy định trong nhiều văn bản quy phạm pháp luật và tài liệu hướng dẫn chuyên môn. Tuy nhiên, chưa có một tài liệu hướng dẫn toàn diện về lĩnh vực này. Với mong muốn thống nhất những nội dung nêu trên trong một tài liệu chính thức, góp phần thúc đẩy hệ thống Cảnh giác dược phát triển, Bộ trưởng Bộ Y tế đã có quyết định thành lập Ban soạn thảo xây dựng Hướng dẫn Quốc gia về Cảnh giác dược bao gồm các thành viên từ các Cục, Vụ chức năng và các đơn vị chuyên môn có liên quan của Bộ Y tế. Hướng dẫn Quốc gia về Cảnh giác dược được xây dựng với mục đích tổng hợp các khái niệm, thuật ngữ quan trọng, xác định vai trò của các thành phần trong hệ thống cũng như hướng dẫn triển khai hoạt động Cảnh giác dược trong một số lĩnh vực chuyên môn cụ thể, đặc biệt là củng cố và phát triển hệ thống báo cáo tự nguyện về phản ứng có hại và về các vấn đề khác liên quan đến tính an toàn của thuốc. Ban biên soạn hy vọng, đây là tài liệu hữu ích dành cho nhân viên y tế, các đơn vị, tổ chức có liên quan để triển khai và phối hợp các hoạt động Cảnh giác dược có hiệu quả, hướng tới đảm bảo sử dụng thuốc hợp lý, an toàn.
Thay mặt Bộ Y tế, tôi xin chân thành cảm ơn các thành viên trong Ban biên soạn Hướng dẫn Quốc gia về Cảnh giác dược, đặc biệt là các cán bộ của Cục Quản lý Dược và Trung tâm Quốc gia về thông tin thuốc và theo dõi phản ứng có hại của thuốc. Sự tâm huyết, nỗ lực của các đồng nghiệp trong Ban biên soạn, cùng với sự góp ý xác đáng của các chuyên gia từ nhiều lĩnh vực trong ngành Y tế cũng như của các đơn vị liên quan đã góp phần quan trọng trong việc xây dựng và hoàn thiện hướng dẫn này.
Trong lần xuất bản đầu tiên, Hướng dẫn khó tránh khỏi sai sót. Chúng tôi rất mong muốn nhận được các ý kiến đóng góp để lần xuất bản sau được hoàn thiện hơn. Mọi ý kiến đóng góp xin gửi về thường trực Ban biên soạn tại Cục quản lý Dược và Trung tâm Quốc gia về thông tin thuốc và theo dõi phản ứng có hại của thuốc.
Xin trân trọng cảm ơn.
GS.TS. Lê Quang Cường
Thứ trưởng Bộ Y tế
MỤC LỤC
| LỜI NÓI ĐẦU | |||
| GIẢI THÍCH THUẬT NGỮ | 1 | ||
| CHƯƠNG 1. TỔNG QUAN VỀ HOẠT ĐỘNG CẢNH GIÁC DƯỢC TẠI VIỆT NAM | 6 | ||
| 1.1. | Cơ sở pháp lý | 7 | |
| 1.2. | Hệ thống Cảnh giác dược tại Việt Nam | 7 | |
| 1.2.1. | Nhiệm vụ của hệ thống Cảnh giác dược Việt Nam | 7 | |
| 1.2.2. | Phạm vi hoạt động | 8 | |
| 1.2.3. | Quy trình hoạt động và vai trò của các thành phần trong hệ thống Cảnh giác dược | 8 | |
| 1.3 | Các phương pháp thu thập thông tin về tính an toàn của thuốc | 13 | |
| 1.3.1. | Phương pháp báo cáo tự nguyện | 13 | |
| 1.3.2. | Phương pháp báo cáo tự nguyện có chủ đích | 14 | |
| 1.3.3. | Phương pháp giám sát chủ động | 14 | |
| 1.4. | Đánh giá nguy cơ/lợi ích của thuốc trong hoạt động quản lý dược phẩm | 15 | |
| 1.4.1. | Cơ sở đánh giá nguy cơ/lợi ích của thuốc tại Việt Nam | 15 | |
| 1.4.2. | Quy trình đánh giá nguy cơ/lợi ích của thuốc ở Việt Nam | 16 | |
| 1.4.3. | Hình thức ra quyết định | 16 | |
| CHƯƠNG 2. HOẠT ĐỘNG CẢNH GIÁC DƯỢC TẠI CÁC CƠ SỞ KHÁM BỆNH, CHỮA BỆNH | 17 | ||
| 2.1. | Giám sát phản ứng có hại của thuốc | 17 | |
| 2.1.1. | Dự phòng | 17 | |
| 2.1.2. | Phát hiện | 18 | |
| 2.1.3. | Xử trí | 19 | |
| 2.1.4. | Đánh giá | 19 | |
| 2.1.5. | Theo dõi phản ứng có hại của thuốc bằng báo cáo tự nguyện | 20 | |
| 2.2. | Giám sát chất lượng thuốc | 23 | |
| 2.2.1. | Một số yếu tố cần xem xét khi phân tích các vấn đề liên quan đến chất lượng thuốc | 23 | |
| 2.2.2. | Báo cáo các vấn đề về chất lượng thuốc | 24 | |
| 2.3. | Giám sát sai sót liên quan đến thuốc (Medication Error, ME) | 24 | |
| 2.3.1. | Một số đặc điểm quan trọng của sai sót liên quan đến thuốc | 24 | |
| 2.3.2. | Các hình thức sai sót liên quan đến thuốc hay gặp | 25 | |
| 2.3.3. | Một số yếu tố gây ra sai sót liên quan đến thuốc thường gặp | 26 | |
| 2.3.4. | Các mức độ nghiêm trọng của hậu quả do sai sót liên quan đến thuốc | 26 | |
| 2.3.5. | Một số biện pháp ngăn chặn các sai sót liên quan đến thuốc | 27 | |
| 2.3.6. | Báo cáo sai sót liên quan đến thuốc | 28 | |
| CHƯƠNG 3. HOẠT ĐỘNG CẢNH GIÁC DƯỢC TRONG SỬ DỤNG THUỐC Y HỌC CỔ TRUYỀN TẠI CÁC CƠ SỞ KHÁM BỆNH, CHỮA BỆNH | 29 | ||
| 3.1. | Giám sát phản ứng có hại của thuốc | 29 | |
| 3.1.1. | Dự phòng | 29 | |
| 3.1.2. | Phát hiện | 32 | |
| 3.2. | Giám sát chất lượng thuốc y học cổ truyền | 33 | |
| 3.2.1. | Phát hiện thuốc y học cổ truyền không đạt tiêu chuẩn chất lượng | 33 | |
| 3.2.2. | Một số yếu tố cần xem xét khi phân tích các vấn đề liên quan đến đảm bảo chất lượng thuốc y học cổ truyền | 35 | |
| 3.2.3. | Xử lý các vấn đề liên quan đến chất lượng thuốc | 36 | |
| 3.3. | Sai sót liên quan đến sử dụng thuốc y học cổ truyền | 36 | |
| 3.3.1. | Các hình thức sai sót liên quan đến thuốc y học cổ truyền | 36 | |
| 3.3.2. | Cách hạn chế sai sót | 37 | |
| 3.4. | Báo cáo phản ứng có hại của thuốc và các vấn đề liên quan đến thuốc | 38 | |
| CHƯƠNG 4. HOẠT ĐỘNG CẢNH GIÁC DƯỢC TRONG CHƯƠNG TRÌNH TIÊM CHỦNG | 39 | ||
| 4.1. | Định nghĩa, phân loại phản ứng sau tiêm chủng | 39 | |
| 4.1.1. | Định nghĩa | 39 | |
| 4.1.2. | Phân loại | 39 | |
| 4.2. | Hướng dẫn giám sát phản ứng sau tiêm chủng | 40 | |
| 4.2.1. | Sơ đồ hệ thống giám sát | 40 | |
| 4.2.2. | Phát hiện, xử trí tai biến nặng sau tiêm chủng | 40 | |
| 4.2.3. | Chế độ báo cáo và quản lý hồ sơ trường hợp phản ứng sau tiêm chủng | 41 | |
| 4.3. | Điều tra tai biến nặng sau tiêm chủng | 43 | |
| 4.3.1. | Thành phần đoàn điều tra | 43 | |
| 4.3.2. | Quy trình điều tra | 43 | |
| 4.3.3. | Lấy mẫu vắc xin để kiểm định | 44 | |
| 4.3.4. | Lấy mẫu bệnh phẩm | 45 | |
| 4.4. | Phân tích kết quả điều tra tai biến nặng sau tiêm chủng | 45 | |
| 4.4.1. | Nhập số liệu theo các biến | 45 | |
| 4.4.2. | Thống kê số liệu | 45 | |
| 4.4.3. | So sánh, đánh giá kết quả | 45 | |
| 4.5. | Đánh giá nguyên nhân tai biến nặng sau tiêm chủng | 45 | |
| 4.5.1. | Các trường hợp cần đánh giá nguyên nhân | 45 | |
| 4.5.2. | Đánh giá nguyên nhân và phân loại các trường hợp tai biến nặng sau tiêm chủng | 45 | |
| CHƯƠNG 5. HOẠT ĐỘNG CẢNH GIÁC DƯỢC TRONG CÁC CHƯƠNG TRÌNH Y TẾ QUỐC GIA | 47 | ||
| 5.1. | Cảnh giác dược trong các chương trình y tế quốc gia | 47 | |
| 5.1.1. | Sự cần thiết triển khai hoạt động Cảnh giác dược | 47 | |
| 5.1.2. | Mối liên quan giữa các chương trình y tế quốc gia với hệ thống Cảnh giác dược | 47 | |
| 5.1.3. | Mục tiêu của Cảnh giác dược trong các chương trình y tế quốc gia | 48 | |
| 5.1.4. | Các phương pháp thu thập thông tin về tính an toàn của thuốc trong các chương trình y tế quốc gia | 48 | |
| 5.2. | Theo dõi phản ứng có hại của thuốc trong các chương trình y tế quốc gia | 49 | |
| 5.2.1. | Chương trình chống Lao Quốc gia | 49 | |
| 5.2.2. | Chương trình phòng, chống HIV/AIDS | 51 | |
| 5.2.3. | Chương trình phòng chống Sốt rét Quốc gia | 53 | |
| CHƯƠNG 6. HOẠT ĐỘNG CẢNH GIÁC DƯỢC TRONG HỆ THỐNG CUNG ỨNG THUỐC | 55 | ||
| A. | Hoạt động Cảnh giác dược tại đơn vị kinh doanh thuốc | 55 | |
| 6.1. | Nhân viên phụ trách hoạt động Cảnh giác dược | 55 | |
| 6.2. | Trách nhiệm của đơn vị kinh doanh thuốc trong thực hành Cảnh giác dược | 56 | |
| 6.2.1. | Báo cáo phản ứng có hại của thuốc | 56 | |
| 6.2.2. | Cập nhật thông tin liên quan đến tính an toàn của thuốc | 57 | |
| 6.2.3. | Lập Kế hoạch quản lý nguy cơ và cập nhật các thay đổi về cân bằng nguy cơ/lợi ích | 58 | |
| B. | Hoạt động Cảnh giác dược tại các cơ sở bán lẻ thuốc | 61 | |
| 6.3. | Các nhà thuốc đạt tiêu chuẩn thực hành tốt nhà thuốc (GPP) | 61 | |
| 6.4. | Các cơ sở bán lẻ khác | 62 | |
| CHƯƠNG 7. THEO DÕI BIẾN CỐ BẤT LỢI CỦA THUỐC TRONG THỬ NGHIỆM LÂM SÀNG | 63 | ||
| 7.1. | Nguyên tắc chung | 63 | |
| 7.2. | Trách nhiệm của các bên trong việc theo dõi và báo cáo biến cố bất lợi trong thử nghiệm lâm sàng tiến hành tại Việt Nam | 63 | |
| 7.2.1. | Nghiên cứu viên chính, nghiên cứu viên tại điểm nghiên cứu | 63 | |
| 7.2.2. | Tổ chức nhận thử, đơn vị triển khai nghiên cứu | 63 | |
| 7.2.3. | Hội đồng Đạo đức/Khoa học cấp cơ sở của tổ chức nhận thử | 63 | |
| 7.2.4. | Nhà tài trợ và các tổ chức được nhà tài trợ ủy quyền | 63 | |
| 7.2.5. | Ban Đánh giá vấn đề đạo đức trong nghiên cứu y sinh học Bộ Y tế | 64 | |
| 7.2.6. | Trung tâm Quốc gia về thông tin thuốc và theo dõi phản ứng có hại của thuốc | 64 | |
| 7.3. | Quy trình, thời hạn và biểu mẫu báo cáo | 64 | |
| 7.3.1. | Báo cáo khẩn cấp | 64 | |
| 7.3.2. | Báo cáo định kỳ | 65 | |
| 7.3.3. | Nơi nhận báo cáo | 65 | |
| 7.3.4. | Hoạt động của các cơ quan liên quan đối với báo cáo AE/SAE | 65 | |
| CHƯƠNG 8. THÔNG TIN THUỐC TRONG HOẠT ĐỘNG CẢNH GIÁC DƯỢC | 66 | ||
| 8.1. | Vai trò của các nguồn dữ liệu tra cứu thông tin thuốc trong hoạt động Cảnh giác dược | 66 | |
| 8.2. | Cập nhật thông tin về an toàn thuốc | 66 | |
| 8.2.1. | Quy trình cập nhật và truyền thông về an toàn thuốc | 66 | |
| 8.2.2. | Các nguồn tài liệu cập nhật thông tin thuốc | 68 | |
| 8.3. | Đánh giá thông tin về phản ứng có hại của thuốc | 69 | |
| 8.3.1. | Quy trình đánh giá thông tin về phản ứng có hại của thuốc | 69 | |
| 8.3.2. | Các nguồn tài liệu cung cấp thông tin về phản ứng có hại của thuốc | 71 | |
DANH MỤC PHỤ LỤC THEO CÁC LĨNH VỰC CỦA HOẠT ĐỘNG CẢNH GIÁC DƯỢC
Phụ lục 1. Mẫu báo cáo phản ứng có hại của thuốc
Phụ lục 2. Mẫu thẻ cảnh báo phản ứng có hại của thuốc
Phụ lục 3. Một số biểu hiện lâm sàng và cận lâm sàng bất thường có thể liên quan đến phản ứng có hại của thuốc
Phụ lục 4. Danh sách một số đối tượng người bệnh và thuốc có nguy cơ cao xuất hiện ADR
Phụ lục 5. Danh sách một số thuốc, xét nghiệm là dấu hiệu gợi ý để phát hiện ADR
Phụ lục 6. Thang đánh giá mối liên quan giữa thuốc và ADR
Phụ lục 7. Mẫu báo cáo bất thường về chất lượng thuốc
Phụ lục 8. Mẫu báo cáo sai sót liên quan đến thuốc
Phụ lục 9. Sơ đồ phân loại sai sót liên quan đến thuốc
Phụ lục 10. Mẫu báo cáo phản ứng có hại của thuốc trong sử dụng thuốc y học cổ truyền
Phụ lục 11. Mẫu báo cáo các trường hợp phản ứng thông thường sau tiêm chủng
Phụ lục 12. Mẫu báo cáo các trường hợp tai biến nặng sau tiêm chủng
Phụ lục 13. Mẫu báo cáo tai biến nặng sau tiêm chủng
Phụ lục 14. Mẫu phiếu điều tra tai biến nặng sau tiêm chủng
Phụ lục 15. Mẫu phiếu lấy và gửi mẫu kiểm định vắc xin
Phụ lục 16. Hướng dẫn lấy mẫu bệnh phẩm đối với các trường hợp tai biến nặng sau tiêm chủng
Phụ lục 17. Biểu mẫu nhập thông tin về tai biến nặng sau tiêm chủng
Phụ lục 18. Bảng tỷ lệ phản ứng sau tiêm chủng của từng loại vắc xin theo Tổ chức Y tế Thế giới
Phụ lục 19. Đánh giá nguyên nhân tai biến nặng sau tiêm chủng
Phụ lục 20. Sơ đồ phân loại nguyên nhân tai biến sau tiêm chủng
Phụ lục 21. Mẫu báo cáo phản ứng có hại của thuốc chống lao sử dụng trong điều trị lao nội trú tại bệnh viện
Phụ lục 22. Mẫu báo cáo phản ứng có hại của thuốc chống lao sử dụng trong điều trị lao tại cộng đồng
Phụ lục 23. Mẫu báo cáo phản ứng có hại của thuốc chống lao sử dụng trong điều trị lao kháng thuốc
Phụ lục 24. Mẫu báo cáo phản ứng có hại của thuốc kháng HIV (ARV)
Phụ lục 25. Mẫu thống kê số lượng báo cáo ADR liên quan đến thuốc ARV tại cơ sở điều trị
Phụ lục 26. Mẫu thống kê số lượng báo cáo ADR liên quan đến thuốc ARV trên địa bàn tỉnh/thành phố
Phụ lục 27. Mẫu báo cáo phản ứng có hại của thuốc trong chương trình phòng chống sốt rét
Phụ lục 28. Mẫu báo cáo CIOMS
Phụ lục 29. Tóm tắt báo cáo định kỳ (PSUR hoặc PBRER)
Phụ lục 30. Mẫu báo cáo an toàn, hiệu quả của thuốc
Phụ lục 31. Mẫu báo cáo tình hình sử dụng thuốc tại cơ sở khám bệnh, chữa bệnh
Phụ lục 32. Mẫu báo cáo biến cố bất lợi nghiêm trọng trong thử nghiệm lâm sàng
Phụ lục 33. Mẫu báo cáo định kỳ biến cố bất lợi trong thử nghiệm lâm sàng
Phụ lục 34. Danh mục văn bản đã ban hành liên quan đến hoạt động Cảnh giác dược
| ADR | Phản ứng có hại của thuốc (Adverse Drug Reaction) |
| AE | Biến cố bất lợi (Adverse Event) |
| CGD | Cảnh giác dược |
| CIOMS | Hội đồng các tổ chức quốc tế về khoa học y học (The Council for International Organizations of Medical Sciences) |
| DĐVN | Dược điển Việt Nam |
| DI | Thông tin thuốc (Drug Infomation) |
| GACP-WHO | Thực hành tốt trồng trọt và thu hái cây thuốc theo hướng dẫn của Tổ chức Y tế Thế giới (World Health Organization Guidelines on Good Agricultural and Collection Practices for Medicinal Plants) |
| GCP | Thực hành lâm sàng tốt (Good Clinical Practice) |
| GMP | Thực hành sản xuất thuốc tốt (Good Manufacturing Practice) |
| GSP | Thực hành bảo quản thuốc tốt (Good Storage Practice) |
| ICH | Hội nghị hòa hợp Quốc tế (International Conference on Harmonisation) |
| NCC MERP | Hội đồng điều phối Quốc gia Hoa Kỳ về báo cáo và phòng tránh sai sót liên quan đến thuốc (National Coordinating Council for Medication Error Reporting and Prevention) |
| OPC | Phòng khám ngoại trú (Out-patient Clinics) |
| PƯSTC | Phản ứng sau tiêm chủng |
| PSUR | Báo cáo định kỳ về tính an toàn của thuốc (Periodic Safety Update Report) |
| PBRER | Báo cáo đánh giá định kỳ về hiệu quả và tính an toàn của thuốc (Periodic Benefit Risk Evaluation Report) |
| QA | Đảm bảo chất lượng (Quality Assurance) |
| QG | Quốc gia |
| RMP | Kế hoạch quản lý nguy cơ (Risk Managemant Plan) |
| SAE | Biến cố bất lợi nghiêm trọng (Serious Adverse Event) |
| SĐK | Số đăng ký |
| SUSAR | Biến cố bất lợi nghiêm trọng ngoài dự kiến (Suspected Unexpected Serious Adverse Reaction) |
| TCMR | Tiêm chủng mở rộng |
| Trung tâm DI&ADR Quốc gia | Trung tâm Quốc gia về thông tin thuốc và theo dõi phản ứng có hại của thuốc |
| VAAC | Cục phòng, chống HIV/AIDS (VietNam Administration of HIV/AIDS Control) |
| UMC | Trung tâm giám sát thuốc toàn cầu Uppsala (Uppsala Monitoring Centre) của Tổ chức Y tế Thế giới |
| WHO | Tổ chức Y tế Thế giới (World Health Organization) |
| YHHĐ | Y học hiện đại |
| YHCT | Y học cổ truyền |
GIẢI THÍCH THUẬT NGỮ
- Báo cáo định kỳ về tính an toàn của thuốc (Periodic Safety Update Report –PSUR) hoặc Báo cáo đánh giá định kỳ về hiệu quả và tính an toàn của thuốc (Periodic Benefit Risk Evaluation Report – PBRER)
Là bản đánh giá cân bằng nguy cơ/lợi ích của một chế phẩm thuốc được thực hiện bởi cơ sở sở hữu số đăng ký của thuốc đó và gửi cho các cơ quan quản lý và cơ quan chuyên môn về dược phẩm tại nước sở tại vào những thời điểm xác định trong chu kỳ sản phẩm giai đoạn hậu mãi. Bản đánh giá này thường được trình bày theo một mẫu chung quốc tế.
- Biến cố bất lợi (adverse event – AE)
Là bất kỳ một biến cố bất lợi nào xảy ra trong quá trình sử dụng thuốc khi điều trị nhưng không nhất thiết là do phác đồ điều trị gây ra.
* Một số văn bản quy phạm pháp luật thuộc các lĩnh vực khác nhau có thể sử dụng định nghĩa khác tương đương (ví dụ: định nghĩa “phản ứng sau tiêm chủng” trong chương trình tiêm chủng).
- Biến cố bất lợi/phản ứng có hại nghiêm trọng (serious adverse event/serious adverse drug reaction – SAE/SADR)
Là các biến cố bất lợi/phản ứng có hại dẫn đến một trong những hậu quả: tử vong; đe dọa tính mạng; buộc người bệnh phải nhập viện để điều trị hoặc kéo dài thời gian nằm viện của người bệnh; để lại di chứng nặng nề hoặc vĩnh viễn cho người bệnh; gây dị tật bẩm sinh ở thai nhi; hoặc bất kỳ phản ứng có hại được nhân viên y tế nhận định là gây ra hậu quả nghiêm trọng về mặt lâm sàng cho người bệnh.
* Một số văn bản quy phạm pháp luật thuộc các lĩnh vực khác nhau có thể sử dụng định nghĩa khác tương đương (ví dụ: định nghĩa “tai biến nặng sau tiêm chủng” trong chương trình tiêm chủng).
- Cảnh giác dược (pharmacovigilance – PV)
Là môn khoa học và hoạt động chuyên môn liên quan đến việc phát hiện, đánh giá, hiểu và phòng tránh biến cố bất lợi hoặc bất kỳ một vấn đề nào khác liên quan đến thuốc.
- Đánh giá nguy cơ/lợi ích (risk/benefit analysis)
Là sự đánh giá tác dụng điều trị tích cực của thuốc so với nguy cơ có thể gặp phải, ví dụ như nguy cơ liên quan đến chất lượng, tính an toàn hoặc hiệu quả điều trị của thuốc đối với sức khỏe người bệnh hoặc sức khỏe cộng đồng.
- Đơn vị kinh doanh thuốc
Trong tài liệu này, đơn vị kinh doanh thuốc bao gồm các công ty đăng ký, sản xuất, xuất nhập khẩu, phân phối thuốc lưu hành tại Việt Nam và các công ty nước ngoài có giấy phép hoạt động về thuốc và nguyên liệu làm thuốc tại Việt Nam.
- Giám sát chủ động (active surveillance)
Là hoạt động bao gồm việc thu thập, phân tích, giải thích và công bố các dữ liệu liên quan đến một hoặc nhiều biến cố bất lợi/phản ứng có hại của thuốc bằng cách sử dụng các phương pháp nghiên cứu quan sát. Việc theo dõi người bệnh được tiến hành chủ động và tất cả các biến cố bất lợi do thuốc xảy ra ngay sau khi bắt đầu điều trị đều được báo cáo một cách thường xuyên, định kì.
- Giảm thiểu nguy cơ (risk minimization)
Là biện pháp can thiệp nhằm ngăn chặn, làm giảm tần suất xuất hiện phản ứng bất lợi hoặc làm giảm mức độ nghiêm trọng của phản ứng có thể xảy ra liên quan đến việc sử dụng một thuốc nào đó.
- Lợi ích (benefit)
Lợi ích của thuốc là tác dụng có lợi ước lượng đạt được khi dùng thuốc đối với một cá thể hoặc quần thể đích.
- Ngày sinh quốc tế của thuốc (international birth date)
Là ngày đầu tiên một hoạt chất có trong chế phẩm thuốc được cấp phép lưu hành dưới dạng bất kỳ một biệt dược nào, của bất kỳ đơn vị kinh doanh thuốc nào và tại bất kỳ quốc gia nào trên thế giới (tham khảo thêm tại http://www.ich.org/).
- Ngày số không (day zero)
Là ngày làm việc đầu tiên mà đơn vị kinh doanh thuốc ghi nhận được thông tin tối thiểu cho một báo cáo đơn lẻ về một biến cố bất lợi của thuốc. Người ghi nhận thông tin có thể là bất kỳ nhân viên nào của đơn vị kinh doanh thuốc hoặc bên thứ ba có thỏa thuận hợp đồng với đơn vị kinh doanh thuốc. Nếu các thông tin tối thiểu về biến cố bất lợi của thuốc được ghi nhận trong phần tóm tắt của một bài báo, y văn thì ngày số không được lấy là ngày tìm kiếm y văn. Đơn vị kinh doanh thuốc nên có biện pháp phù hợp để lấy kịp thời nội dung đầy đủ của bài báo, y văn nhằm xác định tính hợp lệ của một trường hợp.
- Nghiên cứu bệnh chứng (case-control study)
Nghiên cứu bệnh chứng là nghiên cứu trong đó lựa chọn một nhóm cá thể có xuất hiện biến cố và một nhóm khác không xuất hiện biến cố. Mối liên quan giữa thuốc và biến cố xảy ra được kiểm chứng bằng cách so sánh các nhóm này về tiền sử phơi nhiễm với một thuốc có khả năng là nguyên nhân gây ra biến cố.
- Nghiên cứu thuần tập (cohort study)
Nghiên cứu thuần tập là nghiên cứu trong đó lựa chọn một số nhóm cá thể và theo dõi trong một khoảng thời gian để xác định tần suất xuất hiện biến cố. Nghiên cứu thuần tập thường so sánh nhóm có phơi nhiễm với nhóm không phơi nhiễm với thuốc hoặc giữa các bệnh nhân phơi nhiễm với các thuốc khác nhau.
- Nguy cơ (risk)
Nguy cơ của thuốc là bất kỳ tác dụng có hại nào có thể được cho là do thuốc hoặc lo ngại về tác dụng không mong muốn đối với sức khỏe người bệnh, sức khỏe cộng đồng hoặc môi trường.
- Nguy cơ quan trọng (important risk)
Là nguy cơ đã biết hoặc nguy cơ tiềm ẩn có thể ảnh hưởng đến cân bằng nguy cơ/lợi ích của thuốc hoặc ảnh hưởng đến sức khỏe cộng đồng. Nguy cơ được coi là quan trọng phụ thuộc vào một số yếu tố bao gồm đặc điểm cá thể, mức độ nghiêm trọng của nguy cơ và mức độ ảnh hưởng tới sức khỏe cộng đồng. Thông thường, nguy cơ được ghi trong phần chống chỉ định, cảnh báo và thận trọng khi sử dụng của tờ thông tin sản phẩm được coi là quan trọng.
- Nguy cơ tiềm ẩn (potential risk)
Một biến cố bất lợi có cơ sở để nghi ngờ về mối liên quan với thuốc nhưng mối liên quan này chưa được xác thực. Ví dụ:
- Những phát hiện về độc tính ghi nhận được trong các nghiên cứu tiền lâm sàng nhưng chưa được quan sát hoặc được làm sáng tỏ trong các nghiên cứu lâm sàng.
- Biến cố bất lợi ghi nhận được trong các nghiên cứu lâm sàng hoặc các nghiên cứu dịch tễ học trong đó khác biệt so với nhóm chứng (dùng giả dược hoặc hoạt chất, hoặc không dùng thuốc) trên một tham số đối chiếu đủ lớn để đưa ra sự nghi ngờ nhưng chưa đủ lớn để xác định một mối quan hệ nhân quả.
- Một tín hiệu phát sinh từ hệ thống báo cáo tự nguyện các phản ứng bất lợi.
- Một biến cố đã được biết đến là có liên quan với các hoạt chất khác trong cùng một nhóm hoặc có thể được dự đoán là sẽ xảy ra dựa vào đặc tính của thuốc.
- Nguy cơ đã biết (identified risk)
Khi một biến cố bất lợi xảy ra mà có bằng chứng đầy đủ cho thấy có một mối liên quan với thuốc nghi ngờ. Ví dụ:
- Một phản ứng có hại đã được chứng minh đầy đủ trong nghiên cứu tiền lâm sàng và được xác thực bằng các dữ liệu lâm sàng.
- Một biến cố bất lợi được ghi nhận trong các nghiên cứu lâm sàng hoàn chỉnh hoặc các nghiên cứu dịch tễ học mà sự khác biệt so với nhóm chứng trên một tham số đối chiếu đủ lớn để thừa nhận một mối quan hệ nhân quả.
- Một biến cố bất lợi được ghi nhận bởi một số báo cáo tự nguyện đầy đủ trong đó mối quan hệ nhân quả được củng cố chặt chẽ nhờ mối liên hệ về thời gian và sự hợp lý về mặt sinh học, ví dụ sốc phản vệ hoặc phản ứng tại nơi tiêm truyền.
Trong thử nghiệm lâm sàng, nhóm chứng có thể dùng giả dược, hoạt chất hoặc không dùng thuốc.
- Phản ứng có hại của thuốc (adverse drug reaction – ADR)
Theo Luật Dược của Việt Nam (2005), phản ứng có hại của thuốc là những tác dụng không mong muốn có hại đến sức khỏe, có thể xuất hiện ở liều dùng bình thường.
Theo định nghĩa của Tổ chức Y tế thế giới, phản ứng có hại của thuốc là phản ứng độc hại, không mong muốn và xuất hiện ở liều thường dùng cho người với mục đích phòng bệnh, chẩn đoán, điều trị bệnh hoặc làm thay đổi chức năng sinh lý của cơ thể.
* Một số văn bản quy phạm pháp luật thuộc các lĩnh vực khác nhau có thể sử dụng định nghĩa khác tương đương (ví dụ: định nghĩa “phản ứng bất lợi của thuốc” trong thử nghiệm lâm sàng, định nghĩa “tai biến sau tiêm chủng” trong chương trình tiêm chủng).
- Phản ứng có hại ngoài dự kiến (unexpected adverse reaction)
Là các phản ứng có hại có biểu hiện, mức độ nghiêm trọng, tần suất gặp không phù hợp với thông tin kê đơn hay thông tin trên nhãn thuốc.
* Một số văn bản quy phạm pháp luật thuộc các lĩnh vực khác nhau có thể sử dụng định nghĩa khác tương đương (ví dụ: định nghĩa “phản ứng bất lợi của thuốc ngoài dự kiến” trong thử nghiệm lâm sàng).
- Phản ứng có hại nghiêm trọng ngoài dự kiến (suspected unexpected serious adverse reaction – SUSAR)
Là phản ứng có hại ngoài dự kiến và nghiêm trọng, hoặc là các biến cố bất lợi nghiêm trọng, ngoài dự kiến, nghi ngờ liên quan đến thuốc hoặc sản phẩm nghiên cứu.
* Một số văn bản quy phạm pháp luật thuộc các lĩnh vực khác nhau có thể sử dụng định nghĩa khác tương đương (ví dụ: định nghĩa “phản ứng bất lợi nghiêm trọng ngoài dự kiến” trong thử nghiệm lâm sàng).
- Phương pháp báo cáo tự nguyện (spontaneous reporting – SR)
Là phương pháp thu thập các báo cáo riêng lẻ về biến cố bất lợi của thuốc, được nhân viên y tế cũng như các đơn vị kinh doanh dược phẩm báo cáo một cách tự nguyện về Trung tâm Quốc gia và các Trung tâm khu vực về thông tin thuốc và theo dõi phản ứng có hại của thuốc.
- Phương pháp báo cáo tự nguyện có chủ đích (targeted spontaneous reporting – TSR)
Là phương pháp thu thập báo cáo về biến cố bất lợi của thuốc dựa trên nguyên tắc của báo cáo tự nguyện nhưng chỉ tập trung vào việc báo cáo theo một số tiêu chí nhất định như trên một nhóm người bệnh cụ thể, một số phản ứng có hại cụ thể của một số thuốc nhất định. Phương pháp này giữ được các ưu điểm của phương pháp báo cáo tự nguyện (chi phí thấp, dễ áp dụng), đồng thời giúp tập trung vào mục tiêu cụ thể cần theo dõi, nâng cao chất lượng báo cáo và giảm bớt khối lượng công việc cho nhân viên y tế so với báo cáo tự nguyện.
- Quản lý nguy cơ (risk management)
Là những hoạt động Cảnh giác dược và những can thiệp đồng bộ nhằm nhận biết, mô tả, ngăn ngừa và giảm thiểu những nguy cơ liên quan đến thuốc bao gồm cả những biện pháp đánh giá hiệu quả của chính những hoạt động và can thiệp đó.
Mục đích của quản lý nguy cơ là nhằm đảm bảo lợi ích của một thuốc vượt trội so với nguy cơ ở giới hạn cao nhất có thể đạt được cho mỗi cá thể người bệnh cũng như cho toàn bộ nhóm người bệnh đích.
- Sai sót liên quan đến thuốc (medication error – ME)
Sai sót liên quan đến thuốc là bất kỳ biến cố có thể phòng tránh nào có khả năng gây ra hoặc dẫn đến việc sử dụng thuốc không hợp lý, hoặc gây hại cho người bệnh trong khi thuốc được kiểm soát bởi nhân viên y tế, người bệnh, hoặc người sử dụng. Các biến cố như vậy có thể liên quan tới thực hành chuyên môn, các sản phẩm chăm sóc sức khỏe, quy trình và hệ thống bao gồm: kê đơn và quá trình truyền đạt thông tin đơn thuốc; ghi nhãn, đóng gói và danh pháp; pha chế, cấp phát, phân phối; quản lý; giáo dục; giám sát và sử dụng.
- Tác dụng phụ (side effect)
Là tác dụng không mong muốn của một chế phẩm thuốc xảy ra ở liều thường dùng sử dụng ở người và có liên quan đến đặc tính dược lý của thuốc.
- Theo dõi biến cố thuần tập (cohort event monitoring – CEM)
Theo dõi biến cố thuần tập là một nghiên cứu quan sát thuần tập, tiến cứu các biến cố bất lợi liên quan đến một hoặc nhiều loại thuốc. Theo dõi biến cố thuần tập là một quá trình tiến cứu, tiến hành từ khi người bệnh bắt đầu điều trị, biến động (người bệnh mới được bổ sung vào nhóm theo dõi trong quá trình thực hiện chương trình) và theo dõi dọc (các tác dụng được nghiên cứu theo thời gian). Chương trình có những ưu điểm trong việc thu nhận đầy đủ thông tin liên quan đến việc xuất hiện các biến cố và/hoặc phản ứng có hại và có thể tính toán tần suất xảy ra các biến cố.
- Thông tin còn thiếu (missing information)
Là những thông tin về tính an toàn của một thuốc chưa có sẵn tại thời điểm nộp bản Kế hoạch Quản lý Nguy cơ, dẫn đến hạn chế về dữ liệu có thể sử dụng để dự đoán về tính an toàn của thuốc trên thị trường.
- Thử nghiệm lâm sàng (clinical trials)
Thử nghiệm lâm sàng (TNLS) là hoạt động nghiên cứu khoa học một cách hệ thống trên đối tượng nghiên cứu là con người, nhằm đánh giá hiệu quả lâm sàng (hoặc tác dụng dược lý, dược lực học) của sản phẩm nghiên cứu; nhận biết và phát hiện các phản ứng bất lợi; nghiên cứu sự hấp thu, phân bố, chuyển hóa và sự thải trừ của thuốc; nhằm mục đích chứng minh sự an toàn và hiệu quả của sản phẩm thử nghiệm.
Tài liệu này chỉ đề cập đến thử nghiệm lâm sàng thuốc, bao gồm các loại hình nghiên cứu sau: thử nghiệm lâm sàng nhằm chứng minh tính an toàn hoặc hiệu lực/hiệu quả của thuốc trong phát triển thuốc mới và phương pháp điều trị mới, các nghiên cứu trên người về dược động học, sinh khả dụng và tương đương sinh học của thuốc.
- Thuốc giả
Theo Luật Dược của Việt Nam (2005), thuốc giả là sản phẩm được sản xuất dưới dạng thuốc với ý đồ lừa đảo, thuộc một trong những trường hợp sau:
- Không có dược chất;
- Có dược chất nhưng không đúng hàm lượng đã đăng ký;
- Có dược chất khác với dược chất ghi trên nhãn;
- Mạo tên, kiểu dáng công nghiệp của thuốc đã đăng ký bảo hộ sở hữu công nghiệp của cơ sở sản xuất khác.
- Thuốc kém chất lượng
Theo Luật Dược của Việt Nam (2005), thuốc kém chất lượng là thuốc không đạt tiêu chuẩn chất lượng đã đăng ký với cơ quan quản lý.
- Thuốc từ dược liệu
Theo Luật Dược của Việt Nam (2005), thuốc từ dược liệu là thuốc được sản xuất từ nguyên liệu có nguồn gốc tự nhiên từ động vật, thực vật hoặc khoáng chất.
Thuốc có hoạt chất tinh khiết được chiết xuất từ dược liệu, thuốc có sự kết hợp dược liệu với các hoạt chất hóa học tổng hợp không gọi là thuốc từ dược liệu.
- Thuốc y học cổ truyền
Trong tài liệu này, thuốc y học cổ truyền (YHCT) là khái niệm bao gồm dược liệu, vị thuốc y học cổ truyền, thuốc đông y, thuốc từ dược liệu, trong đó:
- Dược liệu là nguyên liệu đạt tiêu chuẩn làm thuốc có nguồn gốc từ thực vật, động vật hay khoáng vật.
- Vị thuốc y học cổ truyền là dược liệu được chế biến, bào chế theo lý luận của y học cổ truyền.
- Thuốc từ dược liệu là thuốc được sản xuất từ nguyên liệu có nguồn gốc tự nhiên từ động vật, thực vật hoặc khoáng chất.
- Thuốc đông y là thuốc từ dược liệu, được bào chế theo lý luận và phương pháp y học cổ truyền của các nước phương Đông.
CHƯƠNG 1: TỔNG QUAN VỀ HOẠT ĐỘNG CẢNH GIÁC DƯỢC TẠI VIỆT NAM
Đảm bảo sử dụng thuốc hợp lý, an toàn và hiệu quả là mục tiêu lớn của ngành y tế. Cùng với các thuốc đã được sử dụng rộng rãi trong lâm sàng, sự ra đời của nhiều thuốc mới đã có những tác động tích cực trong việc kiểm soát bệnh tật và chăm sóc sức khỏe cộng đồng. Bên cạnh đó, những nguy cơ liên quan đến thuốc, đặc biệt là phản ứng có hại của thuốc lại xảy ra khá phổ biến, gây ảnh hưởng không nhỏ tới thành công của điều trị và sức khỏe người bệnh. Phản ứng có hại của thuốc làm nặng thêm tình trạng bệnh, có thể để lại di chứng, đe dọa tính mạng trở thành một trong 10 nguyên nhân gây tử vong hàng đầu tại nhiều quốc gia. Điều đáng lưu ý là đa số phản ứng có hại của thuốc có thể phòng tránh được. Đây chính là cơ sở để xây dựng hệ thống Cảnh giác dược, một hệ thống chuyên biệt của ngành Y tế có nhiệm vụ theo dõi và đánh giá những dữ liệu về tính an toàn của thuốc để ngăn ngừa và giảm thiểu tác động tiêu cực của thuốc đối với người sử dụng, qua đó nâng cao sức khỏe cộng đồng.
Cảnh giác dược (Pharmacovigilance), theo Tổ chức Y tế Thế giới, được định nghĩa là “Môn khoa học và hoạt động chuyên môn liên quan đến việc phát hiện, đánh giá, hiểu và phòng tránh biến cố bất lợi hoặc bất kỳ một vấn đề nào khác liên quan đến thuốc”.
Trước khi đưa ra thị trường, tính an toàn của thuốc, đặc biệt là các phản ứng có hại của thuốc đã được đánh giá qua nhiều giai đoạn nghiên cứu phát triển thuốc (nghiên cứu tiền lâm sàng, thử nghiệm lâm sàng giai đoạn 1, giai đoạn 2 và giai đoạn 3). Tuy nhiên sau khi thuốc được lưu hành, việc sử dụng thuốc không còn bị giới hạn trên một số lượng nhỏ người bệnh và các điều kiện nghiêm ngặt như trong các thử nghiệm lâm sàng mà được mở rộng theo yêu cầu của thực tế điều trị. Lúc này, các vấn đề như đặc điểm người bệnh, bệnh lý mắc kèm, thuốc sử dụng đồng thời, sử dụng thuốc kéo dài, tuân thủ điều trị, … không hoàn toàn giống như trong thử nghiệm lâm sàng. Thêm vào đó, một số vấn đề khác liên quan đến tính an toàn của thuốc như thuốc giả, thuốc kém chất lượng hay sai sót liên quan đến thuốc thường chỉ xuất hiện sau khi thuốc được cấp phép lưu hành và đưa vào sử dụng. Vì vậy, tính an toàn của thuốc vẫn cần được tiếp tục theo dõi và đánh giá sau khi thuốc ra thị trường để có biện pháp can thiệp kịp thời trong trường hợp cần thiết nhằm đảm bảo an toàn cho người bệnh.
Mỗi quốc gia đều cần phát triển hệ thống Cảnh giác dược của mình cho phù hợp với đặc thù về mô hình bệnh tật, yếu tố di truyền, chủng tộc và tình hình sử dụng thuốc. Thực tế điều trị tại nước ta đang đối mặt với nhiều vấn đề trong sử dụng thuốc như lạm dụng thuốc và sử dụng thuốc không hợp lý (lạm dụng kháng sinh, thuốc tiêm và thuốc tiêm truyền, thuốc hỗ trợ, thuốc điều trị triệu chứng, …). Trong quá trình sử dụng thuốc, các sai sót liên quan đến thuốc cũng có thể xảy ra nhưng chưa được đánh giá đầy đủ. Bên cạnh đó, thuốc y học cổ truyền và thuốc có nguồn gốc dược liệu ngày càng được ưu tiên phát triển và sử dụng rộng rãi trong khi vấn đề an toàn khi dùng thuốc chưa thật sự được chú trọng. Các thuốc dự phòng và điều trị lao, HIV/AIDS, sốt rét và các loại vắc xin trong chương trình tiêm chủng được sử dụng cho một số lượng người rất lớn trong cộng đồng nhưng dữ liệu về tính an toàn của các thuốc này trên quần thể người Việt Nam chưa được thu thập và đánh giá một cách có hệ thống. Cảnh giác dược, với mạng lưới theo dõi và các phương pháp thu thập dữ liệu của mình sẽ cung cấp thêm những thông tin quan trọng về tính an toàn của thuốc trong thực tế điều trị, hỗ trợ các cơ quan quản lý nhà nước về y tế trong công tác quản lý sử dụng thuốc, góp phần giảm thiểu tác hại do thuốc gây ra, tiết kiệm chi phí điều trị và đảm bảo an toàn cho sức khỏe cộng đồng.
1.1. Cơ sở pháp lý
Theo điều 51 của Luật Dược (2005), “Bộ Y tế có trách nhiệm tổ chức hệ thống thông tin thuốc và theo dõi phản ứng có hại của thuốc nhằm bảo đảm việc sử dụng thuốc hợp lý, an toàn cho nhân dân; quy định về hoạt động thông tin thuốc tại các cơ sở y tế”; “cơ sở khám bệnh, chữa bệnh, cán bộ, nhân viên y tế có trách nhiệm theo dõi và báo cáo cho người phụ trách cơ sở, cơ quan có thẩm quyền quản lý thuốc về các phản ứng có hại của thuốc”; “trong quá trình lưu hành thuốc, cơ sở sản xuất, phân phối thuốc phải theo dõi, báo cáo cho người phụ trách cơ sở và cơ quan có thẩm quyền quản lý thuốc các phản ứng có hại của thuốc do cơ sở mình sản xuất, phân phối”.
Đề án Quản lý nhà nước về Dược phẩm, An toàn vệ sinh thực phẩm, Mỹ phẩm giai đoạn 2006-2015 ban hành theo Quyết định số 154/2006/QĐ-TTg ngày 30 tháng 06 năm 2006 của Thủ tướng Chính phủ cũng nêu rõ cần “củng cố, kiện toàn và đầu tư nâng cấp về tổ chức, cơ sở vật chất và trang thiết bị cho 2 trung tâm thông tin thuốc và theo dõi phản ứng có hại của thuốc tại thành phố Hà Nội và thành phố Hồ Chí Minh. Thành lập 3 Trung tâm khu vực tại miền núi phía Bắc, Đà Nẵng và Cần Thơ”.
Chiến lược quốc gia phát triển ngành Dược Việt Nam giai đoạn đến năm 2020 và tầm nhìn đến năm 2030 ban hành theo Quyết định số 68/QĐ-TTg ngày 10 tháng 01 năm 2014 của Thủ tướng Chính phủ cũng nhấn mạnh quan điểm “Sử dụng thuốc hợp lý, an toàn, hiệu quả; đẩy mạnh hoạt động Dược lâm sàng và Cảnh giác dược” gắn liền với giải pháp “Tiếp tục hoàn thiện và triển khai tiêu chuẩn thực hành tốt kê đơn thuốc, thực hành tốt nhà thuốc và các chính sách liên quan đến hoạt động Cảnh giác dược, thông tin, quảng cáo thuốc”.
Trên thực tế, hoạt động liên quan đến Cảnh giác dược và giám sát tính an toàn của thuốc đã được triển khai tại Việt Nam từ năm 1994 trong khuôn khổ dự án SIDA “Hỗ trợ hệ thống quản lý Dược” do Chính phủ Thụy Điển tài trợ. Năm 1999, Việt Nam đã trở thành thành viên chính thức của Trung tâm giám sát thuốc toàn cầu Uppsala của Tổ chức Y tế Thế giới (Trung tâm WHO-UMC). Năm 2009, sự ra đời của Trung tâm Quốc gia về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc đã đánh dấu bước ngoặt quan trọng trong hoạt động Cảnh giác dược tại Việt Nam. Năm 2011, Trung tâm khu vực về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc thành phố Hồ Chí Minh trực thuộc Bệnh viện Chợ Rẫy được thành lập. Những đơn vị chuyên môn này cùng với Cục Quản lý Dược, Cục Quản lý Khám, chữa bệnh là đầu mối chính trong hoạt động thông tin thuốc và Cảnh giác dược tại nước ta.
1.2. Hệ thống Cảnh giác dược tại Việt Nam
1.2.1. Nhiệm vụ của hệ thống Cảnh giác dược Việt Nam
(1) Thu thập và quản lý các báo cáo về các vấn đề liên quan đến tính an toàn của thuốc bao gồm: báo cáo phản ứng có hại của thuốc, báo cáo về sai sót liên quan đến thuốc và báo cáo nghi ngờ thuốc giả, thuốc kém chất lượng.
(2) Phối hợp các hoạt động khác liên quan đến thu thập báo cáo về các biến cố bất lợi của thuốc (từ chương trình tiêm chủng và các chương trình y tế quốc gia khác, các thử nghiệm lâm sàng) và các hoạt động giám sát chủ động về biến cố bất lợi của thuốc.
(3) Phát hiện, thông báo kịp thời và xử lý tín hiệu về tính an toàn của thuốc (những biến cố bất lợi chưa biết hoặc chưa được mô tả đầy đủ liên quan đến một thuốc hoặc nhiều thuốc phối hợp).
(4) Cung cấp thông tin về các biến cố bất lợi xảy ra liên quan tới chất lượng thuốc và hỗ trợ công tác quản lý chất lượng thuốc.
(5) Phát hiện và góp phần giảm thiểu các sai sót trong kê đơn, sao chép y lệnh, cấp phát và sử dụng thuốc.
(6) Đánh giá nguy cơ và quản lý nguy cơ liên quan đến thuốc.
(7) Truyền thông có hiệu quả các vấn đề an toàn thuốc bao gồm cả việc bác bỏ những thông tin sai lệch về độc tính của thuốc.
(8) Củng cố và phát triển hoạt động thông tin thuốc. Cập nhật thông tin có được từ hệ thống Cảnh giác dược vào các chính sách thuốc quốc gia, dược thư quốc gia và các hướng dẫn điều trị để mang lại lợi ích cho người bệnh và cộng đồng.
1.2.2. Phạm vi hoạt động
Hoạt động Cảnh giác dược tại Việt Nam hiện nay tập trung vào việc theo dõi các vấn đề liên quan đến tính an toàn của thuốc, bao gồm thuốc hóa dược, vắc xin, sinh phẩm y tế dùng trực tiếp trên người, thuốc y học cổ truyền và thuốc có nguồn gốc dược liệu. Các vấn đề liên quan đến tính an toàn của thuốc bao gồm phản ứng có hại của thuốc, sai sót liên quan đến thuốc, các vấn đề về chất lượng thuốc (thuốc giả, thuốc kém chất lượng). Một số vấn đề khác liên quan đến thuốc như tính an toàn khi sử dụng thực phẩm chức năng và thiết bị y tế không nằm trong phạm vi của hướng dẫn này.
1.2.3. Quy trình hoạt động và vai trò của các thành phần trong hệ thống
Cảnh giác dược Các hoạt động Cảnh giác dược được triển khai theo tiến trình từ báo cáo (gửi thông tin), phát hiện tín hiệu, đánh giá nguy cơ, ra quyết định can thiệp và truyền thông thông tin về tính an toàn của thuốc với sự tham gia của các cá nhân, đơn vị khác nhau trong hệ thống (hình 1). Cụ thể, vai trò của mỗi cá nhân, đơn vị được mô tả như sau:
(1) Người bệnh và cộng đồng
Người bệnh và cộng đồng có trách nhiệm báo cáo cho các nhân viên y tế trong thời gian sớm nhất về bất kỳ một biến cố bất lợi nào xảy ra trong quá trình sử dụng thuốc.
(2) Cơ sở khám bệnh, chữa bệnh
– Lãnh đạo cơ sở khám bệnh, chữa bệnh có trách nhiệm tổ chức và triển khai hoạt động Cảnh giác dược trong đơn vị của mình.
– Nhân viên y tế bao gồm bác sĩ, dược sĩ, điều dưỡng viên, hộ sinh viên, kỹ thuật viên và các nhân viên y tế khác có trách nhiệm theo dõi, phát hiện, xử trí, dự phòng và báo cáo về các phản ứng có hại của thuốc, các sai sót liên quan đến thuốc và các trường hợp nghi ngờ chất lượng thuốc cho người phụ trách về hoạt động Cảnh giác dược trong cơ sở và Trung tâm Quốc gia hoặc Trung tâm khu vực về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc.
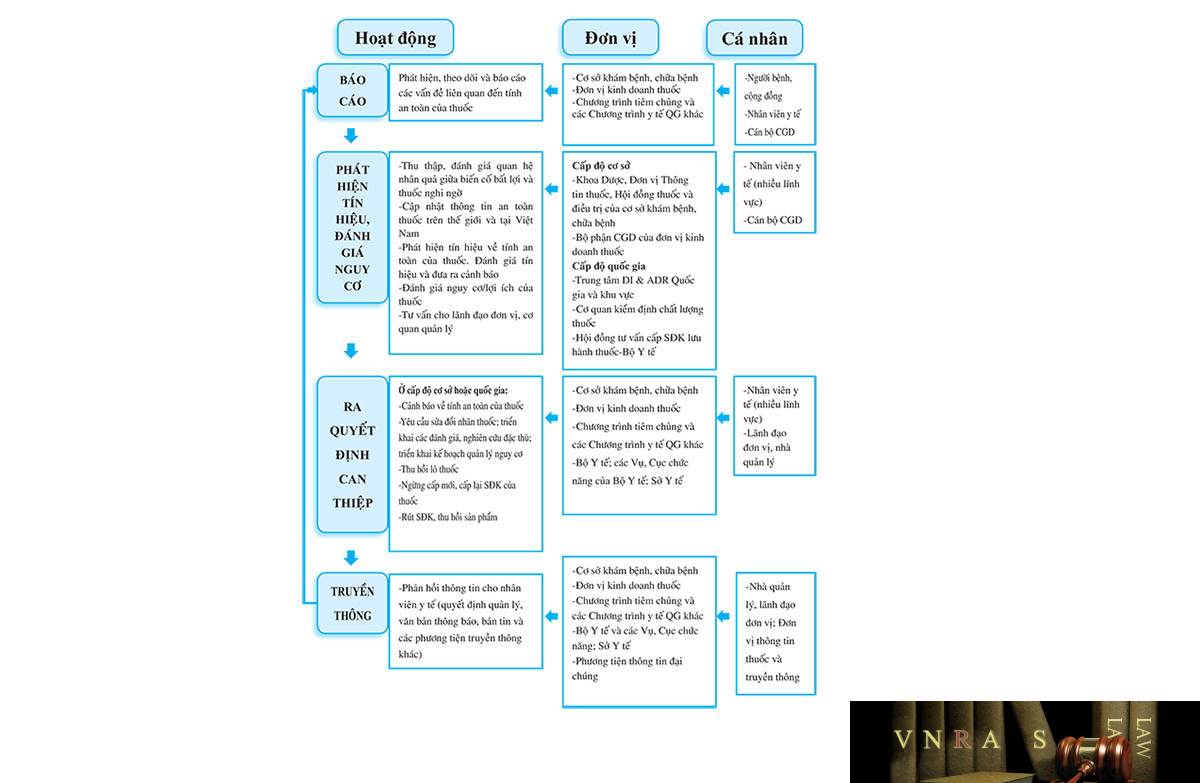
(3) Đơn vị kinh doanh thuốc
Đơn vị kinh doanh thuốc cần thành lập bộ phận chuyên trách về Cảnh giác dược và bố trí cán bộ phụ trách hoạt động này để đảm bảo chất lượng, an toàn của thuốc trong quá trình lưu hành và có biện pháp can thiệp phù hợp trong trường hợp cần thiết. Cụ thể, đơn vị kinh doanh thuốc có trách nhiệm:
– Theo dõi và báo cáo biến cố bất lợi xảy ra trong quá trình lưu hành thuốc do cơ sở mình sản xuất, đăng ký, kinh doanh cho Trung tâm Quốc gia và Trung tâm khu vực về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc theo quy định hiện hành.
– Cập nhật thông tin về chất lượng, an toàn và hiệu quả của thuốc do cơ sở mình sản xuất, đăng ký, kinh doanh cho Cục Quản lý Dược trong trường hợp các thông tin này chưa được cập nhật vào hồ sơ đăng ký thuốc khi thuốc còn đang lưu hành trên thị trường theo quy định hiện hành.
– Trong trường hợp thuốc có số đăng ký (SĐK) lưu hành tại Việt Nam cũng được lưu hành ở các nước khác, cần cập nhật các thay đổi mới về quản lý thuốc liên quan đến vấn đề an toàn như thông tin trên nhãn, hạn chế chỉ định, thu hồi thuốc và rút số đăng ký của cơ quan quản lý dược phẩm nước ngoài cho Cục Quản lý Dược theo quy định hiện hành.
– Xây dựng và triển khai kế hoạch quản lý nguy cơ và giảm thiểu nguy cơ đối với các thuốc có nguy cơ cao do cơ sở mình sản xuất, đăng ký, kinh doanh và khi được Cục Quản lý Dược yêu cầu.
(4) Các cơ sở bán lẻ thuốc
Nhân viên y tế tại các cơ sở bán lẻ thuốc có trách nhiệm ghi nhận phản ánh của người tiêu dùng đối với các biến cố bất lợi xảy ra khi sử dụng thuốc và tham gia hoạt động báo cáo các biến cố nghi ngờ là phản ứng có hại của thuốc cho Trung tâm Quốc gia hoặc Trung tâm khu vực về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc.
(5) Các cơ quan kiểm định chất lượng thuốc
Các cơ quan kiểm định chất lượng thuốc bao gồm Viện Kiểm nghiệm thuốc Trung ương, Viện kiểm nghiệm thuốc thành phố Hồ Chí Minh và các Trung tâm kiểm nghiệm Dược phẩm và Mỹ phẩm tại các tỉnh, thành phố trực thuộc trung ương có trách nhiệm:
– Giám sát chất lượng thuốc theo chức năng, nhiệm vụ được Bộ Y tế giao.
– Lấy mẫu thuốc, kiểm nghiệm, xác định chất lượng thuốc và thông báo kết quả kiểm nghiệm cho Cục Quản lý Dược trong trường hợp cần xác định chất lượng thuốc trong quá trình lưu hành.
(6) Các chương trình y tế quốc gia
Các chương trình y tế quốc gia, đặc biệt là Chương trình chống Lao quốc gia, Chương trình quốc gia phòng chống bệnh Sốt rét, Chương trình phòng chống HIV/AIDS và Chương trình tiêm chủng cần phối hợp với Trung tâm Quốc gia về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc tổ chức và triển khai hoạt động theo dõi phản ứng có hại của thuốc và các vấn đề liên quan đến thuốc trong hệ thống mỗi chương trình theo quy định của Bộ Y tế.
(7) Trung tâm Quốc gia và các Trung tâm khu vực về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc (Trung tâm DI & ADR Quốc gia, Trung tâm DI & ADR khu vực)
Là các cơ quan chuyên môn về thông tin thuốc và Cảnh giác dược, có trách nhiệm:
– Thu thập và quản lý các báo cáo về phản ứng có hại của thuốc, sai sót liên quan đến thuốc và các trường hợp nghi ngờ chất lượng thuốc; tổ chức thẩm định và phản hồi cho người báo cáo.
– Xây dựng và quản lý cơ sở dữ liệu về thông tin thuốc và Cảnh giác dược. – Phát hiện tín hiệu, báo cáo và cung cấp thông tin kịp thời về tính an toàn của thuốc cho các cơ quan quản lý thuộc Bộ Y tế.
– Cung cấp, chia sẻ thông tin cập nhật trong nước và quốc tế về tính an toàn của thuốc cho các tổ chức, cá nhân có liên quan thông qua các hình thức: cổng thông tin điện tử, thư điện tử, ấn phẩm, các phương tiện thông tin đại chúng, … nhằm đảm bảo sử dụng thuốc an toàn, hợp lý trong điều trị và cho cộng đồng.
– Chia sẻ dữ liệu với Trung tâm giám sát thuốc toàn cầu Uppsala của Tổ chức Y tế Thế giới (Trung tâm WHO-UMC), góp phần xây dựng dữ liệu về an toàn thuốc quốc tế.
– Chủ trì hoặc phối hợp triển khai các hoạt động giám sát tích cực, các nghiên cứu về phản ứng có hại của thuốc và các vấn đề liên quan đến thuốc.
– Phát triển hoạt động thông tin thuốc và hướng dẫn chuyên môn cho các đơn vị thông tin thuốc trong các cơ sở khám bệnh, chữa bệnh.
– Tham gia đào tạo, bồi dưỡng nâng cao kiến thức về thông tin thuốc và Cảnh giác dược cho nhân viên y tế, học viên sau đại học, sinh viên y dược.
– Hợp tác và chia sẻ thông tin với các tổ chức quốc tế trong lĩnh vực thông tin thuốc và Cảnh giác dược.
(8) Hội đồng tư vấn cấp số đăng ký lưu hành thuốc của Bộ Y tế
– Tư vấn về chuyên môn, kỹ thuật cho Bộ Y tế trong hoạt động cấp số đăng ký, đánh giá chất lượng, tính an toàn và hiệu quả của thuốc sau khi được cấp số đăng ký.
– Đánh giá tín hiệu về an toàn thuốc và tư vấn cho Bộ Y tế các biện pháp can thiệp về mặt quản lý liên quan đến thuốc trong trường hợp cần thiết.
(9) Các cơ quan quản lý về y tế
Bộ Y tế và các Vụ, Cục chức năng của Bộ Y tế có trách nhiệm:
– Tổ chức hệ thống Cảnh giác dược nhằm bảo đảm việc sử dụng thuốc hợp lý, an toàn.
– Xây dựng, rà soát và ban hành các chính sách y tế có liên quan đến hoạt động Cảnh giác dược.
– Thúc đẩy triển khai, duy trì và đảm bảo về tài chính cho các hoạt động Cảnh giác dược.
Cục Quản lý Dược có trách nhiệm:
– Chủ trì hoặc phối hợp xây dựng, sửa đổi, bổ sung và ban hành theo thẩm quyền các văn bản quy phạm pháp luật, quy định, hướng dẫn chuyên môn kỹ thuật liên quan đến hoạt động Cảnh giác dược.
– Chỉ đạo, quản lý, tổ chức triển khai và giám sát các hoạt động liên quan đến lĩnh vực Cảnh giác dược, theo dõi phản ứng có hại và các thông tin khác liên quan đến thuốc trên phạm vi cả nước, cụ thể bao gồm:
+ Quản lý, thúc đẩy triển khai và giám sát hoạt động Cảnh giác dược trong các đơn vị kinh doanh thuốc tại Việt Nam.
+ Đánh giá nguy cơ/lợi ích và quản lý nguy cơ về tính an toàn của thuốc. + Truyền thông kịp thời, chính xác về các vấn đề an toàn thuốc.
+ Ra quyết định và đảm bảo việc thực hiện quyết định thu hồi, rút số đăng ký thuốc; hạn chế chỉ định, cập nhật thông tin về tính an toàn trên nhãn thuốc; cảnh báo, cung cấp thông tin cho nhân viên y tế và người bệnh về tính an toàn của thuốc.
– Hợp tác và chia sẻ thông tin với các tổ chức quốc tế trong lĩnh vực Cảnh giác dược.
Cục Quản lý Khám, chữa bệnh có trách nhiệm:
– Chủ trì hoặc phối hợp xây dựng, sửa đổi, bổ sung và ban hành theo thẩm quyền các văn bản quy phạm pháp luật, quy định, hướng dẫn chuyên môn kỹ thuật liên quan đến hoạt động Cảnh giác dược trong khám bệnh, chữa bệnh.
– Chỉ đạo, hướng dẫn tổ chức thực hiện và kiểm tra, giám sát việc sử dụng thuốc hợp lý, an toàn và hiệu quả trong khám bệnh, chữa bệnh, trong công tác kê đơn và sử dụng thuốc.
– Chỉ đạo, hướng dẫn tổ chức thực hiện và kiểm tra giám sát thực hành dược lâm sàng; tư vấn, thông tin về sử dụng thuốc và Cảnh giác dược trong các cơ sở khám bệnh, chữa bệnh.
Cục Quản lý Y, Dược cổ truyền có trách nhiệm:
– Chủ trì hoặc phối hợp xây dựng, sửa đổi, bổ sung và ban hành theo thẩm quyền các văn bản quy phạm pháp luật, quy định, hướng dẫn chuyên môn kỹ thuật liên quan đến hoạt động Cảnh giác dược trong sản xuất, chế biến, kinh doanh, kê đơn, cấp phát và sử dụng dược liệu, vị thuốc y học cổ truyền.
– Chỉ đạo, hướng dẫn tổ chức thực hiện và kiểm tra, giám sát việc thực hiện các quy định, hướng dẫn chuyên môn kỹ thuật liên quan đến hoạt động Cảnh giác dược trong sản xuất, chế biến, bảo quản, kinh doanh dược liệu và vị thuốc y học cổ truyền.
– Chỉ đạo, hướng dẫn các cơ sở khám bệnh, chữa bệnh tổ chức thực hiện việc cung ứng và sử dụng hợp lý, an toàn, hiệu quả dược liệu, vị thuốc y học cổ truyền, thuốc đông y, thuốc từ dược liệu bảo đảm chất lượng.
Cục Y tế dự phòng có trách nhiệm:
– Chủ trì hoặc phối hợp với các đơn vị liên quan xây dựng, sửa đổi, bổ sung, ban hành các hướng dẫn chuyên môn kỹ thuật về sử dụng vắc xin, giám sát, xử lý và điều tra nguyên nhân các trường hợp tai biến nặng sau tiêm chủng.
– Chủ trì, phối hợp với các Vụ, Cục liên quan để chỉ đạo, hướng dẫn và kiểm tra việc sử dụng vắc xin trên phạm vi cả nước; thông tin tuyên truyền về an toàn tiêm chủng, lợi ích cũng như các phản ứng có thể gặp sau tiêm chủng.
Cục Khoa học công nghệ và Đào tạo có trách nhiệm:
– Chủ trì hoặc phối hợp xây dựng, sửa đổi, bổ sung, ban hành các hướng dẫn chuyên môn, tiêu chuẩn kỹ thuật cho việc nghiên cứu, phát triển, ứng dụng những sản phẩm y tế mới bảo đảm an toàn, hiệu quả.
– Tổ chức triển khai hệ thống thử nghiệm lâm sàng, Hội đồng đạo đức các cấp, quản lý đảm bảo việc giám sát đầy đủ, kịp thời các biến cố bất lợi xảy ra trong quá trình triển khai thử nghiệm lâm sàng.
– Quản lý và xử lý các báo cáo biến cố bất lợi, báo cáo biến cố bất lợi nghiêm trọng liên quan đến thuốc, sản phẩm y tế trong các thử nghiệm lâm sàng thực hiện tại Việt Nam.
Sở Y tế tỉnh, thành phố trực thuộc trung ương có trách nhiệm:
– Hướng dẫn và tổ chức thực hiện, kiểm tra đánh giá việc thực hiện các văn bản quy phạm pháp luật, các hướng dẫn chuyên môn kỹ thuật về Cảnh giác dược, quản lý chất lượng thuốc và sử dụng thuốc hợp lý, an toàn, hiệu quả tại địa phương theo quy định hiện hành.
– Phổ biến và phối hợp thực hiện các quyết định quản lý của Bộ Y tế và các Vụ, Cục chức năng liên quan đến thu hồi, rút số đăng ký thuốc; cập nhật thông tin về tính an toàn trên nhãn thuốc; cảnh báo, cung cấp thông tin cho nhân viên y tế và người bệnh về tính an toàn của thuốc.
Các đối tác khác trong hệ thống Cảnh giác dược
Hoạt động Cảnh giác dược cũng cần sự tham gia tích cực của các đối tác khác trong hệ thống bao gồm:
– Các cơ sở đào tạo chuyên môn y dược, các viện nghiên cứu chuyên ngành: xây dựng chương trình, tổ chức đào tạo ban đầu, đào tạo lại, bồi dưỡng kiến thức, đào tạo liên tục dành cho nhân viên y tế về thông tin thuốc và Cảnh giác dược, phối hợp triển khai các nghiên cứu trong lĩnh vực Cảnh giác dược.
– Các hội và hiệp hội chuyên môn trong lĩnh vực y dược: cập nhật thông tin liên quan đến tính an toàn của thuốc tại Việt Nam và trên thế giới, tổ chức và phối hợp tổ chức bồi dưỡng chuyên môn, nghiệp vụ về thông tin thuốc và Cảnh giác dược cho hội viên của mình.
– Các tổ chức xã hội và các phương tiện thông tin đại chúng: truyền thông thông tin chính xác và kịp thời về các vấn đề an toàn thuốc cho cộng đồng. – Văn phòng đại diện của Tổ chức Y tế Thế giới tại Việt Nam, Trung tâm giám sát thuốc toàn cầu Uppsala của Tổ chức Y tế Thế giới (Trung tâm WHO-UMC) và các tổ chức phi chính phủ có liên quan: chia sẻ thông tin cập nhật về tính an toàn của thuốc, hỗ trợ chuyên môn kỹ thuật cho các hoạt động Cảnh giác dược tại Việt Nam.
1.3. Các phương pháp thu thập thông tin về tính an toàn của thuốc
Trong lĩnh vực Cảnh giác dược, phương pháp chính được dùng để thu thập thông tin về tính an toàn của thuốc là phương pháp báo cáo tự nguyện. Các phương pháp bổ sung khác có thể được áp dụng trong từng trường hợp cụ thể bao gồm giám sát chủ động và báo cáo tự nguyện có chủ đích.
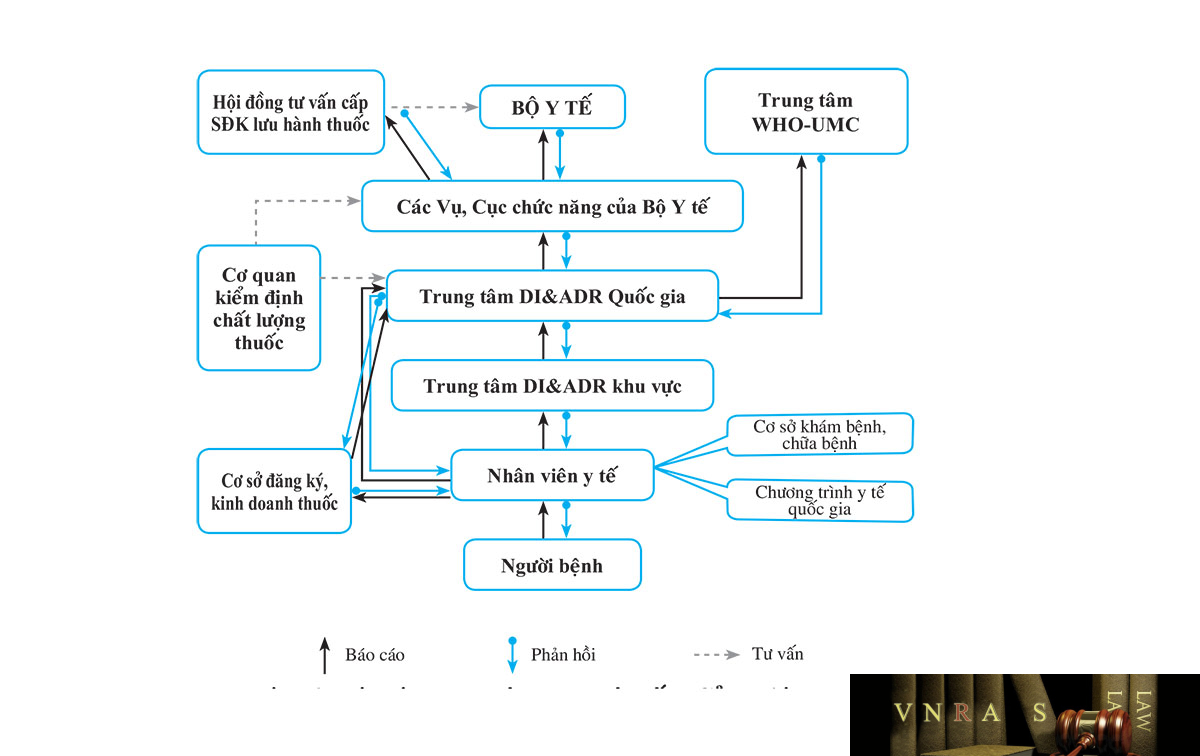
1.3.1. Phương pháp báo cáo tự nguyện
Các báo cáo đơn lẻ về những trường hợp nghi ngờ xảy ra phản ứng có hại của thuốc được nhân viên y tế cũng như các đơn vị kinh doanh dược phẩm báo cáo một cách tự nguyện theo một biểu mẫu báo cáo chung (xem phụ lục 2 của Hướng dẫn này) và gửi về Trung tâm Quốc gia và Trung tâm khu vực về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc (hình 2). Với đặc điểm đơn giản, ít tốn kém, có thể áp dụng với tất cả các thuốc và các loại phản ứng, đây là phương pháp chính được áp dụng để theo dõi phản ứng có hại của thuốc ở tất cả các quốc gia. Thông tin về chất lượng thuốc hay sai sót liên quan đến thuốc cũng có thể được thu thập thông qua hình thức này. Tại các Trung tâm Thông tin thuốc và Theo dõi phản ứng có hại của thuốc, thông tin sẽ được đánh giá, phân tích và phản hồi lại cho người, đơn vị gửi báo cáo. Định kỳ hoặc trong trường hợp phát hiện tín hiệu bất thường liên quan đến tính an toàn của thuốc từ dữ liệu báo cáo tự nguyện, Trung tâm DI & ADR Quốc gia sẽ tổng hợp thông tin và báo cáo với cơ quan quản lý cấp trên (Bộ Y tế và các Vụ, Cục chức năng của Bộ Y tế). Sau khi xin ý kiến tư vấn của các đơn vị chuyên môn khác như Hội đồng tư vấn cấp số đăng ký lưu hành thuốc và các cơ quan kiểm định chất lượng thuốc, Bộ Y tế và các Vụ, Cục chức năng của Bộ Y tế sẽ đưa ra quyết định quản lý kịp thời nhằm đảm bảo an toàn cho người sử dụng thuốc. Bên cạnh đó, dữ liệu mã hóa của báo cáo tự nguyện cũng được chia sẻ với Trung tâm WHO-UMC để đóng góp vào dữ liệu chung về an toàn thuốc toàn cầu.
1.3.2. Phương pháp báo cáo tự nguyện có chủ đích
Báo cáo tự nguyện có chủ đích được dựa trên nguyên tắc của báo cáo tự nguyện. Tuy nhiên, khác với báo cáo tự nguyện, thay vì yêu cầu nhân viên y tế báo cáo về tất cả các phản ứng có hại xảy ra với tất cả các thuốc và mọi đối tượng người bệnh, báo cáo tự nguyện có chủ đích chỉ tập trung theo dõi và báo cáo theo một số tiêu chí nhất định như theo dõi trên một nhóm người bệnh cụ thể, một số phản ứng có hại cụ thể của một số thuốc, phác đồ điều trị. Báo cáo tự nguyện có chủ đích giữ được các ưu điểm của phương pháp báo cáo tự nguyện (chi phí thấp, dễ áp dụng), đồng thời giúp tập trung vào đối tượng cần theo dõi, nâng cao chất lượng báo cáo và giảm bớt khối lượng công việc cho nhân viên y tế so với báo cáo tự nguyện.
1.3.3. Phương pháp giám sát chủ động
Phương pháp này được áp dụng để theo dõi các vấn đề an toàn thuốc trọng tâm thông qua việc chủ động theo dõi người bệnh và ghi nhận tất cả các biến cố bất lợi xảy ra trong quá trình sử dụng thuốc. Việc thu thập báo cáo về biến cố bất lợi của thuốc được thực hiện một cách thường xuyên, định kì từ các cơ sở khám bệnh, chữa bệnh, trung tâm y tế, phòng khám ngoại trú hoặc các cơ sở điều trị được lựa chọn. Biến cố bất lợi được phát hiện bằng cách hỏi trực tiếp người bệnh hoặc theo dõi hồ sơ bệnh án. Giám sát chủ động được thực hiện giống như việc theo dõi dọc trong nghiên cứu dịch tễ học thông qua những bước sau:
– Xây dựng đề cương nghiên cứu trong đó bao gồm mục tiêu, đối tượng, phương pháp nghiên cứu, …
– Thiết kế mẫu thu thập thông tin về người bệnh và mẫu ghi nhận thông tin liên quan đến phản ứng có hại của thuốc. – Tập huấn cho nhân viên y tế tham gia nghiên cứu.
– Triển khai thu nhận người bệnh và theo dõi người bệnh. – Ghi nhận thông tin về biến cố bất lợi tại các lần tái khám.
– Gửi báo cáo cho Trung tâm DI & ADR để tổng hợp, đánh giá và phân tích số liệu.
– Tổng kết, nghiệm thu kết quả và đưa ra kiến nghị.
1.4. Đánh giá nguy cơ/lợi ích của thuốc trong hoạt động quản lý dược phẩm
Đánh giá nguy cơ/lợi ích của thuốc là hoạt động không thể tách rời trong vòng đời của thuốc. Quá trình đánh giá nguy cơ/lợi ích được thực hiện dựa trên thông tin về tính an toàn và hiệu quả của thuốc thu thập trong quá trình phát triển sản phẩm và giám sát hậu mãi sau khi thuốc ra thị trường. Tuy nhiên, quan điểm của các tổ chức, cá nhân trong hệ thống Cảnh giác dược về cân bằng nguy cơ/ lợi ích trong từng trường hợp cụ thể có thể khác nhau. Ở cấp độ quốc gia, cân bằng nguy cơ/lợi ích là yêu cầu đặc biệt quan trọng để một thuốc được duy trì trên thị trường cũng như ảnh hưởng trực tiếp đến phạm vi lưu hành của thuốc. Phần này xin đưa ra những nguyên tắc cơ bản về đánh giá nguy cơ/lợi ích của thuốc có thể áp dụng trong hoạt động quản lý dược phẩm tại Việt Nam.
1.4.1. Cơ sở đánh giá nguy cơ/lợi ích của thuốc tại Việt Nam
Quá trình đánh giá nguy cơ/lợi ích của thuốc được dựa trên những cơ sở sau:
– Thông tin từ cơ sở dữ liệu báo cáo ADR, báo cáo chất lượng thuốc ở Việt Nam.
Trung tâm Quốc gia và Trung tâm khu vực thành phố Hồ Chí Minh về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc là các đơn vị chuyên môn chịu trách nhiệm chính trong việc tiếp nhận các báo cáo phản ứng có hại của thuốc, báo cáo chất lượng thuốc,
phát hiện các tín hiệu cảnh báo về tính an toàn để tư vấn, cung cấp thông tin kịp thời cho cơ quan quản lý của Bộ Y tế.
– Ý kiến tư vấn của các chuyên gia, đơn vị chuyên môn ở Việt Nam.
Ví dụ: các Hội chuyên môn trong lĩnh vực Y-Dược, Hội Bảo vệ sức khỏe người tiêu dùng, Bệnh viện đầu ngành của Bộ Y tế, Trung tâm Quốc gia và Trung tâm khu vực về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc, Trung tâm Dược lý lâm sàng Quốc gia, Trường Đại học Y Hà Nội, …
– Quyết định của các cơ quan quản lý dược phẩm tham chiếu trên thế giới.
Ví dụ: Cơ quan Quản lý Dược phẩm và Thực phẩm Hoa Kỳ (FDA), Cơ quan Quản lý Dược châu u (EMA), Cơ quan Quản lý Y tế Canada (Health Canada), Cơ quan quản lý Dược phẩm và Sản phẩm y tế Anh (MHRA), Cơ quan quản lý Dược phẩm và Sản phẩm y tế Pháp (ANSM), …
– Hướng dẫn chuyên môn của các tổ chức y tế uy tín như Tổ chức Y tế Thế giới (WHO), Hội tim mạch châu u, Hội tim mạch Hoa Kỳ, Hiệp hội đái tháo đường Quốc tế, Hiệp hội đái tháo đường Hoa Kỳ và các hội chuyên môn có uy tín khác trên thế giới.
1.4.2. Quy trình đánh giá nguy cơ/lợi ích của thuốc ở Việt Nam
– Cục Quản lý Dược tiếp nhận thông tin liên quan đến tính an toàn của thuốc từ các nguồn thông tin khác nhau.
– Cục Quản lý Dược tiến hành tổng hợp, xem xét các thông tin liên quan đến thuốc có vấn đề về tính an toàn. Thông tin được tổng hợp dựa trên các căn cứ đã nêu ở phần 1.4.1.
– Cục Quản lý Dược báo cáo, xin ý kiến Hội đồng tư vấn cấp số đăng ký lưu hành thuốc Bộ Y tế (gọi tắt là Hội đồng).
– Hội đồng xem xét, đánh giá, kết luận trên cơ sở các dữ liệu Cục Quản lý Dược cung cấp.
– Cục Quản lý Dược trình Lãnh đạo Bộ Y tế kết luận của Hội đồng.
– Triển khai kết luận của Hội đồng sau khi có ý kiến Lãnh đạo Bộ Y tế.
1.4.3. Hình thức ra quyết định
Căn cứ kết luận của Hội đồng đối với nguy cơ/lợi ích của thuốc và ý kiến của Lãnh đạo Bộ Y tế, Cục Quản lý Dược và Bộ Y tế có thể ra quyết định quản lý theo các mức độ sau:
– Phổ biến, cập nhật thông tin, khuyến cáo cho nhân viên y tế để đảm bảo sử dụng thuốc an toàn, hợp lý.
– Yêu cầu công ty đăng ký, sản xuất thuốc cập nhật thông tin liên quan đến tính an toàn của thuốc trên nhãn, tờ hướng dẫn sử dụng, tờ thông tin cho người bệnh.
– Tạm ngừng cấp số đăng ký mới, đăng ký lại của thuốc; tạm ngừng nhập khẩu nguyên liệu, bán thành phẩm, thành phẩm thuốc.
– Đình chỉ lưu hành thuốc; rút số đăng ký lưu hành thuốc và thu hồi thuốc.
CHƯƠNG 2.
HOẠT ĐỘNG CẢNH GIÁC DƯỢC TẠI CÁC CƠ SỞ KHÁM BỆNH, CHỮA BỆNH
Trong thực hành lâm sàng, phản ứng có hại của thuốc và các vấn đề khác như thuốc giả, thuốc kém chất lượng, sai sót liên quan đến thuốc có tác động tiêu cực tới sức khỏe người bệnh, làm tăng chi phí điều trị, ảnh hưởng lớn đến chất lượng dịch vụ chăm sóc sức khỏe. Các cơ sở khám bệnh, chữa bệnh có trách nhiệm đảm bảo tất cả các loại thuốc được kê đơn và sử dụng cho người bệnh là những thuốc có lợi ích vượt trội nguy cơ và có chất lượng tốt. Hoạt động Cảnh giác dược tại các cơ sở khám bệnh, chữa bệnh bao gồm giám sát (phát hiện, xử trí, báo cáo, đánh giá và dự phòng) các phản ứng có hại của thuốc (ADR) và các vấn đề liên quan đến thuốc trong tất cả các giai đoạn của quá trình quản lý sử dụng thuốc. Những nhiệm vụ này bao gồm:
– Giám sát các phản ứng có hại liên quan đến thuốc hoặc chất lượng thuốc.
– Giám sát các sai sót liên quan đến thuốc.
– Bảo đảm chất lượng thuốc thông qua việc thực hiện tốt các quy định về mua sắm, bảo quản và cấp phát, đồng thời giám sát và giải quyết các vấn đề về chất lượng thuốc.
Đây là trách nhiệm chung của các bác sĩ, y sĩ, dược sĩ, điều dưỡng viên, kỹ thuật viên, hộ sinh viên. Với các cơ sở có các nhân viên y tế khác tham gia công việc liên quan đến khám bệnh, chữa bệnh, việc thực hiện các hoạt động này cần phù hợp chức trách, nhiệm vụ được phân công.
2.1. Giám sát phản ứng có hại của thuốc
2.1.1. Dự phòng
Nhiều phản ứng có hại của thuốc có thể ngăn ngừa được bằng các biện pháp dự phòng trong quá trình sử dụng thuốc cho người bệnh. Phần lớn những phản ứng có hại này là hậu quả của một sai sót liên quan đến thuốc hoặc do thuốc kém chất lượng. Do vậy, có thể áp dụng các biện pháp sau để dự phòng ADR:
a) Nhân viên y tế
– Tuân thủ chỉ định, chống chỉ định, thận trọng, liều dùng của thuốc, chú ý tiền sử dị ứng (thuốc, thức ăn, … ) của bệnh nhân, tương tác thuốc trong kê đơn và thực hiện đầy đủ việc giám sát theo dõi người bệnh trong quá trình điều trị để đảm bảo kê đơn thuốc hợp lý.
– Tuân thủ cảnh báo và thận trọng khi kê đơn sử dụng các thuốc có nguy cơ cao hoặc kê đơn trên đối tượng người bệnh đặc biệt (xem phụ lục 4 của Hướng dẫn này).
– Tuân thủ qui trình bảo quản và sử dụng thuốc cho người bệnh.
– Kiểm tra tương tác thuốc và chống chỉ định trong qui trình cấp phát và sử dụng thuốc.
b) Khoa Dược, Đơn vị Thông tin Thuốc của bệnh viện hoặc bộ phận/người phụ trách công tác Dược tại các đơn vị khám bệnh, chữa bệnh khác
– Cập nhật thông tin sử dụng thuốc, thông tin về thuốc mới, thông tin về an toàn thuốc gửi đến nhân viên y tế và người bệnh trong cơ sở khám bệnh, chữa bệnh dưới nhiều hình thức như tư vấn trực tiếp, sinh hoạt khoa học, sinh hoạt chuyên môn, cung cấp bản tin, tờ thông tin về thuốc.
– Giám sát chất lượng thuốc trước khi cấp phát thuốc về các khoa phòng.
– Hướng dẫn và hỗ trợ nhân viên y tế trong công tác báo cáo ADR.
– Lưu thư cảm ơn đã nhận được báo cáo và phản hồi kết quả thẩm định báo cáo ADR của Trung tâm Quốc gia hoặc Trung tâm khu vực về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc gửi cho nhân viên y tế đã tham gia báo cáo.
c) Hội đồng Thuốc và Điều trị
– Xây dựng qui trình phát hiện, đánh giá, xử trí và báo cáo ADR trong bệnh viện.
– Xác định danh mục các thuốc có nguy cơ cao cần giám sát và xây dựng qui trình hướng dẫn sử dụng các thuốc này trong bệnh viện.
– Tổ chức hội chẩn, thảo luận và đánh giá để đi đến kết luận về hướng xử trí, đề xuất các biện pháp dự phòng phù hợp trong trường hợp xảy ra phản ứng có hại nghiêm trọng tại bệnh viện.
– Định kỳ tổng kết công tác báo cáo ADR trong bệnh viện. Sử dụng các thông tin về tính an toàn để cập nhật, sửa đổi, bổ sung danh mục thuốc của bệnh viện, các hướng dẫn điều trị, qui trình chuyên môn trong bệnh viện.
– Tổ chức tập huấn định kỳ cho nhân viên y tế trong bệnh viện về kỹ năng phát hiện, xử trí, dự phòng ADR, kỹ năng điền báo cáo ADR đúng và đầy đủ thông tin.
2.1.2. Phát hiện
a) Điều dưỡng viên, hộ sinh viên, kỹ thuật viên
– Theo dõi và phát hiện những biểu hiện
lâm sàng và cận lâm sàng bất thường xảy ra trên người bệnh dựa trên các thông tin do người bệnh cung cấp và các triệu chứng ghi nhận được trong quá trình chăm sóc, theo dõi người bệnh (xem phụ lục 3 của Hướng dẫn này).
– Thông báo ngay cho bác sĩ, y sĩ điều trị và khoa Dược (nếu có) về tình trạng bất thường của người bệnh.
– Ghi lại các thông tin liên quan tới các thuốc mà người bệnh đã sử dụng (thuốc nghi ngờ gây ADR và các thuốc dùng đồng thời) bao gồm: tên thuốc, liều dùng, đường dùng, nhà sản xuất, số lô, hạn dùng, ngày và thời gian bắt đầu dùng thuốc, ngày và thời gian kết thúc dùng thuốc (nếu có).
– Giữ lại vỏ bao bì thuốc, vỉ thuốc mà người bệnh đã sử dụng để tham khảo trong trường hợp cần thêm thông tin. Nếu cần thiết, có thể biệt trữ, bảo quản thuốc trong điều kiện bảo quản mà nhà sản xuất khuyến cáo.
b) Bác sĩ, y sĩ
– Phát hiện, ghi nhận lại những biểu hiện lâm sàng và cận lâm sàng bất thường xảy ra trên người bệnh vào bệnh án.
– Kiểm tra lại tất cả các thuốc thực tế người bệnh đã sử dụng.
– Kiểm tra chất lượng cảm quan mẫu thuốc được lưu lại xem có biểu hiện bất thường nào về chất lượng thuốc hay không.
– Kiểm tra lại một số thông tin sau:
+ Lý do sử dụng thuốc, việc sử dụng thuốc có phù hợp tình trạng bệnh lý, có cân nhắc đến các bệnh mắc kèm và chống chỉ định trên người bệnh hay không?
+ Liều dùng thuốc đã phù hợp với liều khuyến cáo chưa? + Người bệnh có tiền sử dị ứng, đặc biệt là dị ứng thuốc không?
+ Có sự phù hợp về thời điểm dùng thuốc nghi ngờ và thời điểm xuất hiện ADR không?
c) Dược sĩ
Trong quá trình thực hiện hoạt động chuyên môn, thông qua xem bệnh án hoặc duyệt thuốc tại khoa Dược, dược sĩ phát hiện ADR dựa trên các thuốc có khả năng được sử dụng để xử trí phản ứng có hại của thuốc, biểu hiện lâm sàng và kết quả xét nghiệm cận lâm sàng bất thường (xem phụ lục 5 của Hướng dẫn này). Ưu tiên xem xét bệnh án của các đối tượng đặc biệt, sử dụng thuốc có nguy cơ cao xảy ra ADR (xem phụ lục 4 của Hướng dẫn này).
2.1.3. Xử trí
a) Điều dưỡng viên, hộ sinh viên, kỹ thuật viên
– Thực hiện xử trí ADR theo đúng y lệnh của bác sĩ điều trị.
– Theo dõi người bệnh và thông báo kịp thời cho bác sĩ điều trị nếu có diễn biến bất thường của người bệnh trong quá trình điều trị tiếp theo.
– Trong trường hợp khẩn cấp, có thể ngừng sử dụng thuốc nghi ngờ gây ảnh hưởng tới tính mạng người bệnh trước khi thông báo cho bác sĩ.
b) Bác sĩ, y sĩ
– Đánh giá mức độ nghiêm trọng của ADR để quyết định hướng xử trí lâm sàng phù hợp.
– Giảm liều hoặc ngừng thuốc nghi ngờ gây ADR trong điều kiện lâm sàng cho phép.
– Kịp thời thực hiện các biện pháp điều trị triệu chứng, điều trị hỗ trợ, đảm bảo chức năng sống cho người bệnh.
– Thực hiện theo các hướng dẫn chuyên môn của Bộ Y tế có liên quan nếu việc xử trí ADR thuộc phạm vi các hướng dẫn đó.
– Trong trường hợp cần thiết, trao đổi hướng xử trí với đồng nghiệp, tổ chức hội chẩn chuyên môn, tham khảo thêm thông tin về ADR từ dược sĩ, Đơn vị Thông tin thuốc của bệnh viện hoặc các Trung tâm Thông tin thuốc và Theo dõi phản ứng có hại của thuốc.
– Giám sát chặt chẽ người bệnh trong trường hợp bắt buộc sử dụng lại thuốc nghi ngờ gây ADR khi không có thuốc thay thế hoặc khi lợi ích của thuốc vượt trội hơn nguy cơ.
c) Dược sĩ
– Trao đổi với bác sĩ điều trị nếu phát hiện ADR khi thực hiện hoạt động dược lâm sàng tại khoa phòng để có biện pháp xử trí phù hợp.
– Cung cấp thông tin về thuốc trong quá trình phát hiện và xử trí ADR theo yêu cầu của nhân viên y tế.
– Hướng dẫn, hỗ trợ bác sĩ và điều dưỡng viên hoàn thiện đầy đủ và chính xác các thông tin cần thiết trong mẫu báo cáo ADR.
– Dược sĩ có thể trực tiếp thu thập thông tin và viết báo cáo ADR.
2.1.4. Đánh giá
a) Đánh giá mối quan hệ nhân quả giữa thuốc và phản ứng có hại
Để đánh giá một biến cố bất lợi có phải do thuốc gây ra hay không, bác sĩ hoặc dược sĩ cần rà soát lại các thông tin liên quan để xem xét nguyên nhân có thể gây ra biến cố, bao gồm:
– Kiểm tra xem người bệnh có dùng đúng thuốc được kê đơn, cấp phát và đúng liều khuyến cáo hay không.
– Hỏi kỹ tiền sử bệnh, tiền sử dị ứng (dị ứng thuốc và các dị ứng khác) để loại trừ các nguyên nhân khác như tình trạng bệnh, thức ăn, các thuốc dùng đồng thời có thể gây ra biến cố.
– Kiểm tra mối liên quan giữa thời gian dùng thuốc và thời điểm xảy ra phản ứng.
– Khám lâm sàng cẩn thận và thực hiện các xét nghiệm liên quan để xác định nguyên nhân gây phản ứng (nếu cần thiết).
– Ghi nhận diễn biến của phản ứng sau khi ngừng thuốc và tái sử dụng thuốc nghi ngờ (nếu có).
– Kiểm tra xem phản ứng đã được ghi nhận trong y văn hay tờ hướng dẫn sử dụng của thuốc nghi ngờ hay không. Những thông tin này giúp củng cố thêm kết luận về mối liên quan giữa thuốc và phản ứng.
– Tùy điều kiện chuyên môn, có thể đánh giá mối liên quan giữa thuốc nghi ngờ và ADR xuất hiện trên người bệnh theo thang phân loại của Tổ chức Y tế Thế giới hoặc thang điểm của Naranjo (xem phụ lục 6 của Hướng dẫn này).
b) Đánh giá mức độ nghiêm trọng của phản ứng có hại
Phản ứng có hại được đánh giá là nghiêm trọng khi các phản ứng này gây ra một trong những hậu quả:
– Trực tiếp hoặc gián tiếp gây tử vong cho người bệnh; – Đe dọa tính mạng người bệnh, cần điều trị cấp cứu; – Người bệnh cần nhập viện hoặc kéo dài thời gian nằm viện; – Người bệnh bị tàn tật vĩnh viễn hoặc nặng nề;
– Dị tật thai nhi;
– Hoặc bất kỳ hậu quả nào của người bệnh mà nhân viên y tế cho là gây hậu quả nghiêm trọng về mặt lâm sàng.
Sau khi đánh giá mối quan hệ nhân quả với thuốc, mức độ nghiêm trọng của phản ứng có hại, nhân viên y tế cần cân nhắc có cần thiết gửi cho người bệnh “Thẻ cảnh báo phản ứng có hại của thuốc” hay không (xem phụ lục 2 của Hướng dẫn này).
Thẻ cảnh báo phản ứng có hại của thuốc là một loại thẻ thông báo cho tất cả nhân viên y tế rằng người mang thẻ đã từng bị phản ứng có hại của thuốc nghiêm trọng. Thẻ này cũng giúp người bệnh biết về những phản ứng nghiêm trọng của họ. Người bệnh cần mang theo thẻ này và đưa cho nhân viên y tế trong tất cả những lần đi khám bệnh. Biện pháp này giúp nhân viên y tế biết được tiền sử bệnh liên quan đến thuốc của người bệnh và giúp tránh phản ứng có hại cùng loại hoặc các phản ứng tương tự.
2.1.5. Theo dõi phản ứng có hại của thuốc bằng báo cáo tự nguyện
Có nhiều phương pháp theo dõi phản ứng có hại của thuốc trong thực hành lâm sàng. Trong khuôn khổ hướng dẫn này, phương pháp báo cáo tự nguyện được giới thiệu do tính chất đơn giản, dễ áp dụng cho các cơ sở khám, chữa bệnh tại Việt Nam.
a) Đối tượng viết báo cáo:
– Người trực tiếp viết báo cáo ADR là bác sĩ, dược sĩ, điều dưỡng viên, hộ sinh viên, kỹ thuật viên và các nhân viên y tế khác.
Khuyến khích nhiều người cùng tham gia viết hoàn thiện báo cáo để nâng cao chất lượng thông tin.
– Thông tin về người báo cáo, người bệnh và đơn vị báo cáo ghi trong phiếu báo cáo phản ứng có hại của thuốc được các Trung tâm về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc bảo mật theo qui định hiện hành.
b) Các trường hợp cần báo cáo
– Báo cáo tất cả các biến cố có hại xảy ra trong quá trình điều trị có nghi ngờ liên quan đến thuốc gây ra bởi:
+ Thuốc, bao gồm cả vắc xin và sinh phẩm y tế, thuốc đông y, thuốc từ dược liệu.
+ Dược liệu, vị thuốc y học cổ truyền.
– Ưu tiên báo cáo:
+ Các phản ứng có hại nghiêm trọng (xem phần 2.1.4 b).
+ Tất cả phản ứng có hại của các thuốc mới đưa vào sử dụng trong điều trị tại bệnh viện.
+ Phản ứng có hại mới chưa từng được biết đến của thuốc (chưa được mô tả trong tờ hướng dẫn sử dụng thuốc, Dược thư Quốc gia Việt Nam, MIMS, Vidal hay các tài liệu tham khảo thông tin thuốc khác).
+ Phản ứng có hại xảy ra liên tục với một thuốc hoặc một lô thuốc trong một thời gian ngắn tại cơ sở khám bệnh, chữa bệnh.
c) Thời gian gửi báo cáo
– Báo cáo cần được gửi trong thời gian sớm nhất có thể sau khi xảy ra phản ứng, ngay cả khi thông tin thu được chưa đầy đủ (báo cáo ban đầu). Trong trường hợp này, có thể bổ sung báo cáo nếu thu thập được thêm thông tin (báo cáo bổ sung).
– Báo cáo trong khi người bệnh chưa xuất viện giúp khai thác đủ thông tin, định hướng làm thêm các xét nghiệm cần thiết để xác định nguyên nhân gây ADR.
– Bảo đảm gửi báo cáo tới Trung tâm Quốc gia hoặc Trung tâm khu vực về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc đúng thời hạn:
+ Báo cáo phản ứng có hại nghiêm trọng gây tử vong hoặc đe dọa tính mạng người bệnh: gửi trong thời gian sớm nhất có thể nhưng không muộn hơn 7 ngày làm việc kể từ thời điểm xảy ra phản ứng.
+ Báo cáo phản ứng có hại nghiêm trọng còn lại: gửi trong thời gian sớm nhất có thể nhưng không muộn hơn 15 ngày làm việc kể từ thời điểm xảy ra phản ứng.
+ Báo cáo phản ứng có hại không nghiêm trọng có thể tập hợp gửi hàng tháng, trước ngày mùng 5 của tháng kế tiếp.
d) Hướng dẫn điền mẫu báo cáo phản ứng có hại của thuốc
– Nguyên tắc chung:
+ Hoàn thành mẫu báo cáo với đầy đủ thông tin nhất có được từ bệnh án.
+ Sử dụng một bản báo cáo riêng cho mỗi người bệnh.
+ Trường hợp dùng thuốc để điều trị ADR nhưng lại gây ra một ADR khác cho người bệnh nên tách thành một báo cáo riêng.
+ Chữ viết rõ ràng, viết chính xác tên thuốc, hạn chế viết tắt.
+ Điền thông tin chính xác, thống nhất, tránh mâu thuẫn giữa các thông tin trong báo cáo.
– Mẫu báo cáo ADR: sử dụng mẫu báo cáo phản ứng có hại của thuốc theo quy định hiện hành (xem phụ lục 1 của Hướng dẫn này).
– Các thông tin tối thiểu cần điền trong mẫu báo cáo ADR + Thông tin về người bệnh: họ và tên, tuổi, giới tính.
+ Thông tin về phản ứng có hại: mô tả chi tiết biểu hiện ADR, ngày xuất hiện phản ứng, diễn biến ADR sau khi xử trí (bao gồm diễn biến sau khi ngừng thuốc hoặc giảm liều thuốc hoặc tái sử dụng thuốc nghi ngờ).
+ Thông tin về thuốc nghi ngờ: tên thuốc nghi ngờ, liều dùng, đường dùng, lý do dùng thuốc, ngày và thời điểm bắt đầu dùng thuốc.
+ Thông tin về người và đơn vị báo cáo: tên đơn vị báo cáo, họ và tên người báo cáo, chức vụ, số điện thoại liên lạc hoặc địa chỉ email (nếu có).
Với các thông tin còn lại trong mẫu báo cáo, khuyến khích nhân viên y tế thu thập, bổ sung tối đa thông tin.
– Hướng dẫn chi tiết các thông tin cần điền trong báo cáo ADR: nhân viên y tế điền mẫu báo cáo theo quy định của Bộ Y tế và theo hướng dẫn của Trung tâm Quốc gia về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc tại trang thông tin điện tử http:// canhgiacduoc.org.vn/.
e) Hình thức gửi báo cáo ADR
– Với các cơ sở khám bệnh, chữa bệnh có khoa Dược: nhân viên y tế gửi báo cáo ADR tới khoa Dược là đầu mối tập hợp báo cáo trong bệnh viện. Trong trường hợp khẩn cấp, có thể gửi báo cáo trực tiếp đến Trung tâm Quốc gia hoặc Trung tâm khu vực về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc, sau đó thông báo lại cho khoa Dược.
– Với các cơ sở khám bệnh, chữa bệnh không có khoa Dược: nhân viên y tế gửi báo cáo trực tiếp đến Trung tâm Quốc gia hoặc Trung tâm khu vực về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc.
– Báo cáo ADR được điền vào mẫu báo cáo theo qui định và gửi về Trung tâm Quốc gia hoặc các Trung tâm khu vực về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc bằng một trong 5 hình thức sau:
+ Cách 1: gửi qua bưu điện.
+ Cách 2: gửi qua thư điện tử (email).
+ Cách 3: báo cáo ADR trực tuyến. Truy cập vào trang web: http://baocaoadr.vn/. Đọc và làm theo hướng dẫn trên trang web.
+ Cách 4: gửi qua fax.
+ Cách 5: điện thoại báo cáo trực tiếp cho Trung tâm trong trường hợp rất khẩn cấp. Thông tin sau đó cần được điền vào mẫu báo cáo và gửi về Trung tâm theo một trong 4 cách nêu trên.
f) Nơi nhận báo cáo
Báo cáo có thể gửi về một trong hai địa chỉ sau:
– Trung tâm Quốc gia về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc (nhận báo cáo từ tất cả các tỉnh/thành phố trên phạm vi toàn quốc)
Địa chỉ: Trường Đại học Dược Hà Nội, 13-15 Lê Thánh Tông, Quận Hoàn Kiếm, Hà Nội
Điện thoại: (04) 3933 5618
Fax: (04) 3933 5642
E-mail: di.pvcenter@gmail.com
Trang thông tin điện tử: http://canhgiacduoc.org.vn
– Trung tâm khu vực về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc thành phố Hồ Chí Minh (nhận báo cáo của các tỉnh/thành phố từ Đà Nẵng trở vào)
Địa chỉ: Bệnh viện Chợ Rẫy, 201B Nguyễn Chí Thanh, Phường 12, Quận 5, Thành phố Hồ Chí Minh
Điện thoại: (08) 3855 4137- Ext: 794 hoặc (08) 3856 3537 Fax: (08) 3856 3537
E-mail: adrhcm@choray.vn
g) Xử lý báo cáo về phản ứng có hại của thuốc tại Trung tâm Quốc gia và Trung tâm khu vực về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc
– Qui trình tiếp nhận báo cáo:
+ Khi nhận được báo cáo ADR, Trung tâm Quốc gia hoặc Trung tâm khu vực sẽ gửi thư xác nhận cảm ơn tới đơn vị/cá nhân đã gửi báo cáo.
+ Các báo cáo ADR sẽ được Trung tâm Quốc gia hoặc Trung tâm khu vực thẩm định theo quy trình xử lý báo cáo ADR của Trung tâm.
– Qui trình đánh giá và phản hồi:
+ Định kỳ hàng năm, Trung tâm Quốc gia tổ chức tổng kết, phân loại báo cáo ADR, gửi báo cáo tổng kết công tác báo cáo ADR về Bộ Y tế, Sở Y tế các tỉnh, thành phố trực thuộc Trung ương, Y tế ngành và các cơ sở khám bệnh, chữa bệnh.
+ Trong trường hợp cần phản hồi nhanh, đặc biệt với các ADR nghiêm trọng, Trung tâm Quốc gia và Trung tâm khu vực sẽ nhanh chóng tiến hành thu thập thông tin và thẩm định để gửi phản hồi cho nhân viên y tế, cơ sở khám bệnh, chữa bệnh đã gửi báo cáo.
2.2. Giám sát chất lượng thuốc
Một nhiệm vụ quan trọng trong các cơ sở khám bệnh, chữa bệnh là giám sát và xem xét các báo cáo về thuốc kém chất lượng. Các dấu hiệu về chất lượng thuốc kém có thể biểu hiện dưới các hình thức sau:
– Hình thức của thuốc không đảm bảo khi quan sát bằng mắt thường, theo báo cáo của nhân viên y tế, ví dụ như thay đổi màu sắc, vỡ, rò rỉ, có mùi lạ, …
– Thuốc có hiệu quả điều trị kém.
– Thuốc gây ra phản ứng có hại.
2.2.1. Một số yếu tố cần xem xét khi phân tích các vấn đề liên quan đến chất lượng thuốc
– Các loại thuốc không ổn định hoặc có chỉ số điều trị hẹp có thể gặp nhiều vấn đề về chất lượng hơn các loại thuốc khác.
– Các sản phẩm cùng một hoạt chất được sản xuất bởi các nhà sản xuất khác nhau có thể có sự khác biệt về sinh khả dụng, do đó không tương đương sinh học và tương đương điều trị.
– Có sự khác biệt về độ ổn định của các dạng bào chế khác nhau của các loại thuốc uống. Nói chung, các dạng bào chế thể rắn ổn định hơn các dạng lỏng, đặc biệt là trong điều kiện nhiệt độ hoặc độ ẩm cao.
Khi có báo cáo về chất lượng thuốc, bệnh viện cần đánh giá xem đây có phải là do có sai sót ở một trong các khâu như sản xuất (bao gồm cả thuốc giả), bảo quản, cấp phát, quản lý, sử dụng thuốc, … hay không. Quá trình này có thể bao gồm các bước sau:
– Xác định chính xác bản chất của vấn đề về chất lượng thuốc.
– Kiểm tra cảm quan về sản phẩm, bao gồm cả xác định thời hạn sử dụng, bao bì và ghi nhãn.
– Tìm thêm thông tin về quá trình mua sắm, bảo quản và phân phối của sản phẩm.
– Quan sát cách thức sản phẩm đã được sử dụng, ví dụ như kỹ thuật pha chế, tiêm thuốc, quá trình cấp phát. Trong trường hợp cần thiết có thể phỏng vấn người bệnh để kiểm tra sự tuân thủ.
– Quan sát xem người bệnh được điều trị như thế nào để loại trừ các nguyên nhân khác như sai sót liên quan đến thuốc, tương tác thuốc, …
– Báo cáo tới cơ quan quản lý cấp trên về việc phát hiện dấu hiệu thuốc có chất lượng kém.
2.2.2. Báo cáo các vấn đề về chất lượng thuốc
Khuyến khích nhân viên y tế và các cơ sở khám bệnh, chữa bệnh báo cáo các vấn đề về chất lượng thuốc gặp phải trong quá trình điều trị.
Nhân viên y tế nên báo cáo các vấn đề về chất lượng thuốc khi gặp phải các trường hợp sau:
– Nghi ngờ thay đổi tính chất hóa, lý của thuốc như: + Thuốc thay đổi màu;
+ Thuốc bị tách lớp/tách các thành phần, có kết tủa;
+ Bị biến thành bột hoặc vỡ vụn;
+ Đóng cứng;
+ Thay đổi mùi.
– Nghi ngờ có vấn đề về độ ổn định (ví dụ như những bất thường về tính chất lý hóa của thuốc trong bảo quản, pha chế, sử dụng, …).
– Bao bì hoặc ghi nhãn sai hoặc không đầy đủ.
– Nghi ngờ nhiễm vi sinh vật.
– Các thành phần của sản phẩm thuốc bị lỗi.
– Thất bại điều trị.
– Thuốc hết hạn.
Các cơ sở khám bệnh, chữa bệnh có thể tham khảo mẫu báo cáo bất thường chất lượng thuốc tại phụ lục 7 của tài liệu này để ghi nhận thông tin và gửi báo cáo tới cơ quan quản lý cấp trên, Trung tâm Quốc gia hoặc Trung tâm khu vực về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc.
2.3. Giám sát sai sót liên quan đến thuốc (Medication Error, ME)
2.3.1. Một số đặc điểm quan trọng của sai sót liên quan đến thuốc
– Sai sót liên quan đến thuốc có thể phòng tránh được. Đây là đặc điểm quan trọng nhất và vì vậy, việc ngăn chặn và giảm thiểu sai sót liên quan đến thuốc trở thành một mục tiêu quan trọng trong chính sách an toàn thuốc của mỗi Quốc gia cũng như của các cơ sở y tế.
– Sai sót liên quan đến thuốc có thể gây ra bởi những sai sót trong giai đoạn lên kế hoạch dùng thuốc hoặc giai đoạn thực hiện kế hoạch. Như vậy sai sót liên quan đến thuốc có thể xảy ra ở bất kỳ giai đoạn nào và việc hạn chế sai sót phải mang tính chất hệ thống.
– Sai sót liên quan đến thuốc bao gồm cả sai sót do quên thuốc (quên liều hoặc kê đơn thiếu thuốc) hoặc sai sót do sử dụng sai.
– Sai sót liên quan đến thuốc có thể đã gây hại cho người bệnh hoặc chưa (chưa tính đến việc gây ra hậu quả bất lợi trên người bệnh). Ví dụ như bác sĩ kê đơn liều thuốc chưa phù hợp với hướng dẫn nhưng ngay sau đó được dược sĩ thông báo và có sửa lại liều đúng thì vẫn xác định là đã có sai sót xảy ra.
2.3.2. Các hình thức sai sót liên quan đến thuốc hay gặp
– Sai người bệnh: sai sót xảy ra khi đưa thuốc cho không đúng người bệnh.
– Sai thuốc: sai sót do dùng thuốc không theo đúng chỉ định cho người bệnh.
– Sai liều: bao gồm quá liều, thấp hơn liều điều trị, quên liều, hoặc đưa thêm liều không đúng chỉ định hoặc không nhớ liều dùng cho người bệnh.
– Sai đường dùng.
– Sai thời điểm dùng: quá sớm, quá muộn, nhầm lẫn việc dùng thuốc trước hay sau bữa ăn hoặc không rõ thời gian dùng.
– Sai tốc độ truyền: tốc độ truyền quá nhanh, quá chậm hoặc không rõ tốc độ truyền.
– Sai khoảng thời gian dùng thuốc: người bệnh dùng thuốc với khoảng cách giữa các lần dùng ngắn hơn hoặc dài hơn khoảng thời gian được chỉ định.
– Sai dạng bào chế, hàm lượng hoặc nồng độ của thuốc.
– Sai kỹ thuật dùng thuốc: như dùng sai dung môi pha thuốc, bẻ, nghiền thuốc không được phép nghiền.
– Dùng thuốc hết hạn hoặc đã bị hư hỏng do điều kiện bảo quản.
– Dùng thuốc cho người bệnh có tiền sử bị dị ứng với thuốc đó.
– Dùng thuốc có chống chỉ định cho người bệnh do tương tác giữa thuốc-bệnh lý đang có của người bệnh, tương tác thuốc-thuốc, tương tác thuốc-thức ăn.
– Người bệnh có hành vi không đúng: người bệnh tự dùng thuốc hoặc thực phẩm chức năng có tương tác với thuốc điều trị, kể cả người bệnh đã biết hoặc không biết phải tránh dùng đồng thời các thuốc này; người bệnh không tuân thủ chỉ định và hướng dẫn sử dụng thuốc.
Sai sót liên quan đến thuốc có thể gặp ở bất kỳ giai đoạn nào trong quá trình sử dụng thuốc từ khi kê đơn đến giai đoạn giám sát bệnh nhân trong quá trình điều trị. Do vậy, cũng có thể phân loại sai sót theo giai đoạn sử dụng thuốc như sau:
– Sai sót trong giai đoạn kê đơn: sai sót khi kê đơn thuốc, thường gặp là chọn thuốc không hợp lý, kê đơn không phù hợp, không hiệu quả (không đúng số lượng, liều dùng, nồng độ, hàm lượng, số lần dùng thuốc, đường dùng, hoặc hướng dẫn sử dụng), kê đơn thiếu thuốc hoặc thừa thuốc, lỗi viết đơn thuốc, bao gồm cả chữ viết khó đọc.
– Sai sót trong giai đoạn sao chép đơn thuốc: sai về tên thuốc, dạng bào chế, đường dùng, liều dùng, chế độ liều, thiếu thuốc, thuốc không được kê đơn.
– Sai sót trong giai đoạn cấp phát: cấp phát thuốc không được kê đơn, cấp phát sai liều dùng, thiếu thuốc, sai dạng bào chế (thường gặp với các các thuốc có hình thức tương tự nhau, tên đọc giống nhau).
– Sai sót trong giai đoạn dùng thuốc: sai kỹ thuật dùng, thời gian dùng, dùng thuốc không được kê đơn cho người bệnh, sai liều dùng, thiếu liều, không đối chiếu nhận dạng thuốc.
– Sai sót trong quá trình giám sát: không thay đổi thuốc, không hiệu chỉnh liều khi cần thiết hoặc điều chỉnh sai thuốc, không theo dõi nồng độ điều trị với các thuốc có yêu cầu theo dõi điều trị.
2.3.3. Một số yếu tố gây ra sai sót liên quan đến thuốc thường gặp
– Cường độ làm việc cao và tình trạng mệt mỏi của nhân viên y tế.
– Nhân viên thiếu kinh nghiệm và được đào tạo chưa đầy đủ.
– Truyền đạt thông tin không hiệu quả giữa các nhân viên y tế (bao gồm cả chữ viết tay khó đọc và y lệnh bằng lời) và giữa nhân viên y tế với người bệnh.
– Các yếu tố môi trường, ví dụ như ánh sáng yếu, nhiều tiếng ồn, bị gián đoạn công việc thường xuyên.
– Tăng chủng loại hoặc số lượng thuốc sử dụng cho một bệnh nhân.
– Số lượng và mức độ phức tạp của các phép tính phải thực hiện để kê đơn, cấp phát hoặc sử dụng thuốc (ví dụ đổi đơn vị, tính nồng độ dung dịch tiêm, tốc độ truyền, tính liều theo cân nặng, theo diện tích da, …).
– Nhiều loại thuốc trong danh mục và một số dạng bào chế (ví dụ như thuốc tiêm) có liên quan đến gia tăng sai sót.
– Nhầm lẫn về danh pháp, bao bì hoặc nhãn mác.
– Thiếu các chính sách và quy trình hiệu quả.
2.3.4. Mức độ nghiêm trọng của hậu quả lâm sàng do sai sót liên quan đến thuốc
Hội đồng điều phối Quốc gia Hoa Kỳ về báo cáo và phòng tránh sai sót liên quan đến thuốc (National Coordinating Council for Medication Error Reporting and Prevention, NCC MERP) đã đề xuất phương pháp phân loại theo mức độ nghiêm trọng của hậu quả lâm sàng trên bệnh nhân nhằm đánh giá tác động của sai sót liên quan đến thuốc tới bệnh nhân. Theo phương pháp này, sai sót thuốc được phân vào 9 nhóm từ A đến I theo mức độ nghiêm trọng của hậu quả lâm sàng trong bảng 1.
Bảng 1. Phân loại sai sót liên quan đến thuốc theo mức độ nghiêm trọng của hậu quả lâm sàng trên bệnh nhân
| Không có sai sót | Nhóm A | Tình huống hoặc biến cố có khả năng gây ra sai sót |
| Sai sót, không gây tổn hại | Nhóm B | Có sai sót nhưng thuốc chưa tiếp cận đến bệnh nhân. |
| Nhóm C | Có sai sót và đã tiếp cận đến bệnh nhân nhưng không gây tác hại. | |
| Nhóm D | Sai sót xảy ra và đã tiếp cận đến bệnh nhân dẫn đến yêu cầu cần theo dõi để xác định không gây tác hại cho bệnh nhân và/hoặc cần can thiệp để dự phòng/giảm thiểu tác hại cho bệnh nhân. | |
| Sai sót, gây tổn hại | Nhóm E | Sai sót xảy ra có thể đã gây tác hại hoặc góp phần gây tác hại tạm thời cho bệnh nhân và cần các biện pháp can thiệp. |
| Nhóm F | Sai sót xảy ra có thể đã gây tác hại hoặc góp phần gây tác hại tạm thời cho bệnh nhân và cần nhập viện hoặc kéo dài thời gian nằm viện. | |
| Nhóm G | Sai sót xảy ra có thể đã gây tác hại hoặc góp phần gây tác hại không hồi phục cho bệnh nhân. | |
| Nhóm H | Sai sót xảy ra yêu cầu các can thiệp cần thiết để duy trì sự sống. | |
| Sai sót, gây tử vong | Nhóm I | Sai sót xảy ra góp phần hoặc gây ra gây ra tử vong cho bệnh nhân. |
Quy trình để xác định mức độ nghiêm trọng của hậu quả lâm sàng do sai sót gây ra được trình bày chi tiết ở phụ lục 9 của tài liệu này.
2.3.5. Một số biện pháp ngăn chặn các sai sót liên quan đến thuốc
– Thành lập nhóm chuyên môn gồm bác sĩ, điều dưỡng và dược sĩ để xây dựng các tiêu chuẩn thực hành lâm sàng phù hợp với điều kiện của cơ sở.
– Thiết lập hệ thống thu thập và ghi nhận thông tin về sai sót liên quan đến thuốc tại cơ sở. Hệ thống này cần xây dựng trên cơ sở không quy kết để khuyến khích nhân viên y tế báo cáo sai sót.
– Xây dựng các quy trình kèm theo các hướng dẫn và bảng kiểm để quản lý việc sử dụng các dịch truyền tĩnh mạch và các loại thuốc có nguy cơ cao như insulin, các thuốc chống đông, thuốc gây nghiện, … (xem phụ lục 4 của Hướng dẫn này).
a) Khi kê đơn thuốc
Các cơ sở khám bệnh, chữa bệnh cần yêu cầu các thầy thuốc áp dụng hướng dẫn Thực hành kê đơn tốt khi kê đơn thuốc cho người bệnh, trong đó cần lưu ý những nội dung sau:
– Tuân thủ các quy định về kê đơn và hướng dẫn sử dụng thuốc, biết rõ các thông tin cần ghi trong đơn thuốc đặc biệt là thông tin về người bệnh (kể cả tiền sử dị ứng thuốc) và thông tin về thuốc. Khi kê đơn thuốc cần lưu ý về dạng bào chế của thuốc (trong đó cần chú ý dạng giải phóng đặc biệt); về những thuốc có tên tương tự nhau hoặc nghe tương tự nhau; về mục đích dùng thuốc và về quy trình sử dụng thuốc tại đơn vị.
– Chuẩn hóa kê đơn và cách truyền đạt thông tin đơn thuốc giữa các nhân viên y tế trong quy trình điều trị. Không khuyến cáo việc kê đơn thuốc bằng lời (ví dụ qua điện thoại) trong mọi trường hợp.
– Kê đơn bằng cách viết tay phải rõ ràng để tránh nhầm lẫn, hạn chế những chữ viết tắt, viết đơn vị và liều dùng, tính liều rõ ràng trước khi đơn được chuyển đi.
b) Khi cấp phát thuốc
– Nhân viên y tế (dược sĩ và điều dưỡng) cần tuân thủ các quy định chuyên môn về cấp phát thuốc. Đặc biệt lưu ý việc đối chiếu so sánh nội dung trên bao bì của thuốc với thông tin trong đơn kê hoặc phiếu lĩnh thuốc, đối chiếu nhãn thuốc, đơn kê và người bệnh.
– Tìm kiếm và phân loại riêng những thuốc có thể gây ra những sai sót nghiêm trọng tại bệnh viện.
– Xây dựng và thực hiện nghiêm ngặt quy trình bảo quản thuốc.
– Sử dụng các công cụ ghi nhớ như thiết kế nhãn phụ hoặc dùng phần mềm nhắc nhở trên máy tính để hạn chế sự nhầm lẫn khi cấp phát các thuốc có tên nhìn tương tự nhau, bao bì có màu sắc giống nhau.
– Giữ nguyên nhãn mác, y lệnh, bao bì ban đầu của thuốc trong suốt quá trình cấp phát hoặc có bao bì phù hợp đối với các thuốc cấp phát lẻ không còn nguyên bao gói.
c) Khi sử dụng thuốc cho người bệnh
– Nhân viên y tế (dược sĩ và điều dưỡng) cần tuân thủ các quy định chuyên môn về sử dụng thuốc cho người bệnh. Đặc biệt lưu ý việc xác định đúng người bệnh khi đưa thuốc và giám sát chặt chẽ người bệnh sau khi dùng thuốc.
– Phiên giải và thực hiện đúng các thông tin trong đơn thuốc. Đặc biệt lưu ý đơn thuốc được viết bằng tay, có chữ viết tắt, kê đơn không hoàn chỉnh hay kê đơn bằng lời.
– Lưu ý những thuốc có tên tương tự nhau khi nghe và khi viết, thuốc có bao bì gần giống nhau.
– Bảo quản thuốc phù hợp, đặc biệt các thuốc có nồng độ đậm đặc, thuốc có yêu cầu bảo quản đặc biệt.
2.3.6. Báo cáo sai sót liên quan đến thuốc
Khi có sai sót xảy ra, nhân viên y tế cần báo cáo với bộ phận phụ trách có liên quan tại cơ sở khám bệnh, chữa bệnh (ví dụ như một tiểu ban giám sát ADR và sai sót trong điều trị). Tiểu ban giám sát ADR và sai sót trong điều trị sẽ thu thập thông tin và giúp Hội đồng thuốc và điều trị của bệnh viện tổ chức hội chẩn, thảo luận và đánh giá để đi đến kết luận cho hướng xử trí và đề xuất các biện pháp dự phòng tiếp theo. Việc phân loại hậu quả những sai sót liên quan đến thuốc có thể tham khảo tại phụ lục 9 của Hướng dẫn này. Những thông tin về sai sót cũng cần được thông báo hoặc tập huấn để rút kinh nghiệm trong cơ sở, đồng thời, có thể cần cập nhật, bổ sung, sửa đổi danh mục thuốc, hướng dẫn điều trị và các qui trình chuyên môn khác tại cơ sở để phòng tránh các sai sót.
Khuyến khích nhân viên y tế và các cơ sở khám bệnh, chữa bệnh báo cáo tất cả các sai sót liên quan đến thuốc gặp phải trong quá trình điều trị về Trung tâm Quốc gia về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc (xem phụ lục 8 của Hướng dẫn này).
CHƯƠNG 3.
HOẠT ĐỘNG CẢNH GIÁC DƯỢC TRONG SỬ DỤNG THUỐC Y HỌC CỔ TRUYỀN TẠI CÁC CƠ SỞ KHÁM BỆNH, CHỮA BỆNH
Hiện nay, ở nhiều nước trên thế giới cũng như tại Việt Nam vẫn tồn tại song hành hai hình thức chữa bệnh bằng y học hiện đại (YHHĐ) và y học cổ truyền (YHCT). Thuốc YHCT được sử dụng lâu đời trong hệ thống khám bệnh, chữa bệnh và được xem là ít tác dụng không mong muốn. Tuy nhiên, trong thực tế sử dụng, các hiện tượng dị ứng, ngộ độc có liên quan đến thuốc YHCT cũng đã được ghi nhận. Hiện tại có rất ít báo cáo chính thức hay nghiên cứu cụ thể về phản ứng có hại liên quan đến sử dụng thuốc YHCT. Do vậy, việc triển khai các hoạt động Cảnh giác dược đối với thuốc YHCT cũng như tiến hành các nghiên cứu sâu hơn về dược liệu cổ truyền có nguy cơ gây hại là rất cần thiết. Những thông tin, dữ liệu thu được từ các hoạt động này sẽ là cơ sở để đưa ra cảnh báo, khuyến cáo sử dụng thuốc YHCT an toàn, hợp lý, tạo được niềm tin cho người dân và phát huy thế mạnh của YHCT trong phòng và chữa bệnh.
Thuốc YHCT bao gồm dược liệu, vị thuốc y học cổ truyền, thuốc đông y, thuốc từ dược liệu, trong đó:
– Dược liệu là nguyên liệu đạt tiêu chuẩn làm thuốc có nguồn gốc từ thực vật, động vật hay khoáng vật.
– Vị thuốc y học cổ truyền là dược liệu được chế biến, bào chế theo lý luận của y học cổ truyền.
– Thuốc từ dược liệu là thuốc được sản xuất từ nguyên liệu có nguồn gốc tự nhiên từ động vật, thực vật hoặc khoáng chất.
– Thuốc đông y là thuốc từ dược liệu, được bào chế theo lý luận và phương pháp y học cổ truyền của các nước phương Đông.
Thuốc YHCT thường được sử dụng dưới nhiều dạng: thuốc sắc uống, thuốc ngâm rượu (uống, dùng ngoài), cồn thuốc, thuốc bột, cao thuốc (cao đặc, cao lỏng, cao xoa, cao dán), chè thuốc, thuốc cốm, thuốc viên (viên nang, viên nén, viên hoàn,…). Thuốc YHCT có thể là một vị thuốc (dược liệu sơ chế hoặc đã chế biến) hoặc một chế phẩm thuốc được phối ngũ lập phương và bào chế theo phương pháp YHCT để điều trị bệnh theo lý luận của YHCT.
Hoạt động Cảnh giác dược trong các cơ sở có sử dụng thuốc YHCT tuân thủ theo hướng dẫn chung trong các cơ sở khám bệnh, chữa bệnh đồng thời cần tính đến đặc thù của thuốc YHCT. Những hoạt động này bao gồm:
– Giám sát các phản ứng có hại, có thể do hoạt chất thuốc hoặc có thể do sai sót liên quan đến thuốc hoặc chất lượng thuốc kém.
– Giám sát các sai sót liên quan đến thuốc.
– Bảo đảm chất lượng thuốc thông qua việc thực hành tốt về mua sắm, bảo quản và cấp phát, đồng thời giám sát và giải quyết các vấn đề về chất lượng thuốc.
3.1. Giám sát phản ứng có hại của thuốc
3.1.1. Dự phòng
Để hạn chế những phản ứng có hại do dùng thuốc YHCT gây ra, các cơ sở khám chữa bệnh YHCT cần tuân thủ những nguyên tắc sau:
a) Đối với thầy thuốc YHCT
Tuân thủ chỉ định, chống chỉ định, thận trọng, liều dùng của thuốc, chú ý tiền sử dị ứng của bệnh nhân (dị ứng thuốc, thức ăn, …), tương tác thuốc trong kê đơn (giữa các thuốc YHCT hoặc giữa thuốc YHCT và thuốc tân dược). Thực hiện đầy đủ việc giám sát theo dõi người bệnh trong quá trình điều trị để đảm bảo kê đơn thuốc hợp lý.
Thận trọng khi kê đơn sử dụng các thuốc có nguy cơ cao (vị thuốc có độc tính, có dược tính mạnh, vị thuốc dễ bị ngụy tạo) hoặc kê đơn trên đối tượng người bệnh đặc biệt.
Khi kê đơn thuốc cần lưu ý:
– Kê đơn phải phù hợp tình trạng bệnh
Theo YHCT, bệnh tật sinh ra do mất sự cân bằng âm dương trong cơ thể. Khi kê đơn, người thầy thuốc cần chú ý đến vị trí nông sâu của bệnh (biểu – lý), tính chất của bệnh (hàn – nhiệt) và trạng thái người bệnh (hư – thực). Một chứng bệnh cũng chia ra các thể khác nhau như thể hàn, thể nhiệt, thể hư (bản thân các cơ quan trong cơ thể bị hư suy), thể thực (bệnh cấp tính do yếu tố bên ngoài là chủ yếu chưa ảnh hưởng tới công năng của các tạng trong cơ thể), … Mỗi một thể có phương pháp điều trị khác nhau, trong đó cần lưu ý: tránh dùng những thuốc thuộc nhóm giải biểu cho những bệnh nhân tự hãn hoặc đạo hãn, bệnh nhân xuất huyết, thiếu máu, …; tránh dùng thuốc thuộc nhóm thanh nhiệt cho bệnh nhân khi bệnh tà còn ở biểu, tỳ vị hư nhược, hiện tượng giả nhiệt, …; tránh dùng các thuốc thanh nhiệt hóa đàm cho những người dương hư; tránh dùng thuốc ôn hóa hàn đàm cho những người âm hư do có thể gây mất tân dịch.
Tuy không có phương thuốc chung cho mọi bệnh nhưng cần đảm bảo nguyên tắc: bệnh hàn thì dùng thuốc nhiệt, bệnh nhiệt thì dùng thuốc hàn, hư nên dùng thuốc bổ, bệnh thực phải dùng thuốc tả để công phạt, … Nếu dùng trái đi có thể gây hậu quả nghiêm trọng: bệnh thể nhiệt mà dùng thuốc cay nóng, bệnh thuộc hàn mà dùng thuốc lạnh đều làm cho bệnh nặng thêm.
– Lưu ý khi phối ngũ các vị thuốc
Mục đích của việc phối ngũ kết hợp các vị thuốc trong YHCT nhằm tăng hiệu quả điều trị, đồng thời làm giảm các tác dụng không mong muốn. Các loại phối ngũ trong y học cổ truyền bao gồm: tương tu, tương sử, tương úy, tương sát, tương ố, tương phản. Phối ngũ tương úy (kiềm chế lẫn nhau) dùng vị thuốc này để hạn chế tác dụng có hại của vị thuốc kia (ví dụ: Bán hạ sống gây ngứa dùng với Gừng tươi cho hết ngứa, như vậy bán hạ sống tương úy với Gừng tươi). Trong phối ngũ tương sát (tiêu trừ độc tính của nhau), vị thuốc này có thể làm mất độc tính của vị thuốc kia (ví dụ phòng phong trừ độc thạch tín; đậu xanh trừ độc ba đậu. Có thể vận dụng tương sát để giải độc khi ngộ độc asen hoặc ba đậu). Hai vị thuốc tương ố là khi dùng chung sẽ làm giảm tác dụng của nhau như Hoàng cầm với
Sinh khương. Hai vị thuốc tương phản khi kết hợp có thể làm tăng độc tính của nhau. Các vị thuốc tương phản lẫn nhau bao gồm: Cam thảo phản Cam toại, Nguyên hoa, Hải tảo; Ô đầu phản Bối mẫu, Bán hạ, Bạch cập, Bạch tiễn.
– Lưu ý, kiêng kị khi dùng thuốc
+ Không dùng thức ăn chống lại tác dụng của thuốc, ví dụ không ăn đồ ăn lạnh khi dùng thuốc ôn trung khu hàn (nóng, ấm); không nên ăn chất béo, nhờn, tanh khi dùng các thuốc kiện tỳ tiêu tích; không nên ăn uống chất kích thích khi dùng thuốc an thần; kiêng ăn thịt lợn khi thang thuốc có các vị thuốc như Cam thảo, Hoàng liên, Cát cánh, Ô mai; kiêng ăn dấm khi dùng Bạc hà.
+ Với thuốc dùng ngoài, cần cảnh báo bệnh nhân không được dùng đường uống.
+ Không dùng thuốc quá liều hoặc kéo dài. Một số nhóm thuốc như thuốc hành khí, hoạt huyết, phá huyết không được dùng liều cao hoặc kéo dài do dễ làm hao tổn chính khí và tổn thương tân dịch. Một số vị thuốc YHCT khi dùng quá liều trong một thời gian dài có thể gây tổn hại cho cơ thể. Ví dụ: Mộc thông là vị phổ biến để lợi tiểu, dùng với liều cao kéo dài có thể gây nên suy thận. Dùng liều cao Tế tân, Bạch quả, Ô đầu, Phụ tử, Đào nhân cũng có thể gây ngộ độc. Dùng kéo dài Chu sa, Đại giả thạch (những vị thuốc có nguồn gốc khoáng vật) hoặc Lục thần khúc (một vị thuốc tăng cường kích thích tiêu hóa) có thể ảnh hưởng tới chức năng gan và thận.
+ Chỉ dẫn cách sắc thuốc trong đơn theo đúng quy định.
+ Thận trọng sử dụng thuốc YHCT cho các đối tượng phụ nữ có thai, trẻ em, người cao tuổi. Ví dụ: cấm kỵ dùng các vị thuốc có tác dụng trục thủy, tả hạ phá khí, phá huyết hoặc thận trọng đối với các vị thuốc tả hạ, hoạt huyết, đại nhiệt, … cho phụ nữ có thai; không dùng thuốc chỉ khái (chữa ho) cho trẻ em khi bị sởi lúc bắt đầu mọc ban hay đang mọc ban do có thể ảnh hưởng tới việc mọc ban và dễ gây biến chứng; không dùng thuốc tả hạ cho người cao tuổi dương hư suy yếu.
b) Khoa Dược, khoa YHCT, Đơn vị Thông tin Thuốc của bệnh viện hoặc bộ phận/người phụ trách công tác Dược tại các đơn vị khám chữa bệnh khác
Hoạt động Cảnh giác dược tại khoa Dược, khoa YHCT, Đơn vị Thông tin Thuốc của bệnh viện hoặc bộ phận/người phụ trách công tác Dược tại các đơn vị khám bệnh, chữa bệnh khác cần tuân thủ theo hướng dẫn chung trong chương 2 và lưu ý một số nội dung cụ thể sau:
– Bộ phận cấp phát: khi tiếp nhận thuốc do các khoa phòng chuyển xuống, người phụ trách bộ phận phải kiểm tra đơn thuốc trước khi đưa vào cân, báo cho thầy thuốc khi đơn thuốc bị thiếu vị thuốc, tuân thủ theo chỉ định của thầy thuốc đối với những vị thuốc đặc biệt cần gói riêng, sắc riêng trước khi cho người bệnh uống. Khi cân chia thuốc, cần đảm bảo cân đủ, chia đều vào các thang; không tự ý thay vị thuốc bị thiếu trong đơn. Trên thang thuốc phải ghi rõ tên người bệnh, số khoa phòng, số giường (đối với bệnh nhân nội trú). Đối với bệnh nhân ngoại trú, phải dặn dò bệnh nhân cách sắc thuốc, tuân thủ theo chỉ định của thầy thuốc trước khi cấp thuốc cho bệnh nhân mang thuốc về sắc.
– Bộ phận sắc thuốc: tuân thủ theo đúng quy trình sắc thuốc theo quy định hiện hành của Bộ Y tế (quy trình số 71 ban hành kèm theo Quyết định 26/2008/QĐ-BYT của Bộ Y tế ngày 22 tháng 7 năm 2008 về quy trình kỹ thuật y học cổ truyền), lưu bã thuốc theo quy định hiện hành (Thông tư 05/2014/TT-BYT của Bộ Y tế ban hành ngày 14 tháng 02 năm 2014 quy định việc sử dụng dược liệu, vị thuốc y học cổ truyền trong các cơ sở khám bệnh, chữa bệnh).
– Bộ phận bào chế: đảm bảo bào chế theo phương pháp của Bộ Y tế quy định (Quyết định 39/2008/QĐ-BYT của Bộ Y tế ban hành ngày 15 tháng 12 năm 2008 về phương pháp chung chế biến các vị thuốc theo phương pháp cổ truyền, Quyết định 3759/QĐ-BYT của Bộ Y tế ban hành ngày 08 tháng 10 năm 2010 về phương pháp chế biến đảm bảo chất lượng của 85 vị thuốc đông y và Quyết định 3635/QĐ-BYT của Bộ Y tế ngày 16 tháng 09 năm 2014 về phương pháp chế biến đảm bảo chất lượng đối với 18 vị thuốc đông y. Các quyết định này là cơ sở thống nhất công tác bào chế, chế biến thuốc y học cổ truyền nhằm đảm bảo chất lượng thuốc YHCT khi đưa vào sử dụng. Đối với cơ sở không có điều kiện chế biến, khoa Dược cần yêu cầu cơ sở cung ứng phải chế biến trước khi cấp phát sử dụng cho bệnh nhân (yêu cầu cụ thể kèm theo theo hợp đồng cung ứng). Trong trường hợp chế biến thuốc tại bệnh viện, cần xây dựng phương pháp bào chế, chế biến và tiêu chuẩn cơ sở của vị thuốc, chế phẩm sản xuất tại cơ sở và được người đứng đầu cơ sở phê duyệt để sử dụng trong cơ sở khám bệnh, chữa bệnh.
– Bộ phận kiểm nhập: việc kiểm tra chất lượng đối với dược liệu và vị thuốc tại các cơ sở khám bệnh, chữa bệnh hiện chủ yếu dựa vào cảm quan, vì vậy cán bộ kiểm nhập phải là người có chuyên môn và kinh nghiệm thực tế để nhận biết các dược liệu, vị thuốc đúng, đảm bảo chất lượng trước khi nhập kho. Trong trường hợp nghi ngờ chất lượng thuốc YHCT, cần yêu cầu đổi lại hoặc báo cho hội đồng kiểm nhập để xem xét quyết định đưa đi kiểm nghiệm. Thêm vào đó, phiếu kiểm nghiệm cần thể hiện đầy đủ chỉ tiêu theo quy định hiện hành.
– Kho bảo quản: bảo quản thuốc theo quy định, định kỳ kiểm tra, kiểm kê, không để thuốc mốc, mối, mọt hoặc hết hạn sử dụng.
– Thường xuyên cung cấp thông tin về an toàn thuốc và xây dựng bộ mẫu dược liệu đối chiếu để nhân viên y tế tham khảo.
c) Hội đồng Thuốc và Điều trị
– Xây dựng tiêu chí lựa chọn, tiêu chuẩn cơ sở thuốc YHCT sử dụng trong bệnh viện theo đúng quy định và đáp ứng với nhu cầu sử dụng tại cơ sở.
– Xác định danh mục các vị thuốc có nguy cơ giả mạo cao, dễ nhầm lẫn để tăng cường kiểm tra, giám sát trong bệnh viện.
– Tăng cường giám sát việc thực hiện quy chế kê đơn, quy định về sử dụng thuốc an toàn và hiệu quả trong bệnh viện.
d) Người bệnh
Để ngăn ngừa những tai biến do dùng thuốc YHCT, người bệnh cần phải tuân thủ triệt để hướng dẫn của thầy thuốc, không tùy tiện sử dụng thuốc YHCT khi không có chỉ định, không tự ý nâng liều và kéo dài thời gian sử dụng. Một số vị thuốc và bài thuốc đơn giản theo kinh nghiệm dân gian có thể tự dùng, nhưng tốt nhất vẫn nên có sự tư vấn đầy đủ của thầy thuốc chuyên khoa. Khi sử dụng, nếu thấy bất kỳ dấu hiệu bất thường nào thì phải ngừng thuốc ngay và báo lại cho thầy thuốc biết để xử trí kịp thời.
Người bệnh có nhu cầu điều trị bệnh bằng YHCT nên đến các bệnh viện YHCT, các khoa YHCT trong hệ thống y tế công hoặc các cơ sở YHCT được cấp phép hoạt động để được điều trị và được hướng dẫn sử dụng thuốc một cách an toàn, hiệu quả.
3.1.2. Phát hiện
Phản ứng có hại khi sử dụng thuốc YHCT có thể xảy ra từ từ hoặc có thể tiến triển cấp tính. Một số phản ứng đã được ghi nhận khi sử dụng thuốc YHCT bao gồm:
– Các biểu hiện dị ứng trên da từ nhẹ đến nặng như mày đay, ban đỏ, phù Quincke, hội chứng Lyell, hội chứng Stevens-Johnson.
– Các biểu hiện ngộ độc thuốc như: ăn kém, đau bụng, ỉa chảy, nôn, buồn nôn co giật, sốt, đau rát, vàng da, mệt mỏi, co giật, tê lưỡi, khó thở, mạch nhanh, huyết áp hạ, trụy tim mạch.
– Các biểu hiện tại các cơ quan khác như tuần hoàn, hô hấp, tiêu hóa, thận, … cũng đã được ghi nhận. Trong đó có các triệu chứng cấp tính như đau rát, tiểu ít, sưng miệng, khô miệng, đại tiện lỏng hoặc táo bón, đầy bụng, sôi bụng, … Ngoài ra, một số phản ứng có hại có thể tiến triển từ từ sau một thời gian dài sử dụng thuốc YHCT như suy tim, suy gan, suy thận hoặc ung thư khi dùng kéo dài các thuốc có chứa chất độc đối với tim, gan, thận.
Cần phân biệt mộtsố tác dụng có hại có thể xảy ra do trộn thuốc tân dược vào thuốc YHCT như: loét dạ dày, gây xốp xương, phù, tăng huyết áp, rậm lông (nếu trong thuốc có các hoạt chất corticosteroid), loét đường tiêu hóa, xuất huyết, dị ứng (đối với thuốc chống viêm không steroid), buồn ngủ, khô miệng (đối với cyproheptadin), suy gan (đối với paracetamol); nhức đầu, chóng mặt, khó chịu ở dạ dày (đối với sildenafil), tụt huyết áp, trụy tim mạch (đối với các dẫn chất nitrat như nitroglycerin, isosorbid dinitrat), …
Trong nhiều trường hợp, việc xác định thuốc y học cổ truyền gây ra phản ứng rất khó khăn. Khi nghi ngờ một biến cố là phản ứng có hại của thuốc YHCT, người thầy thuốc cần lưu ý:
– Mô tả lại phản ứng một cách rõ ràng.
– Tìm hiểu tiền sử bệnh nhân để loại trừ tất cả những nguyên nhân có thể giải thích cho biến cố đó như các bệnh mắc kèm, thức ăn, các thuốc tân dược dùng phối hợp với thuốc YHCT, hoặc các vị thuốc phối ngũ trong đơn thuốc có khả năng gây ra tương tác thuốc. Chú ý đến mối quan hệ thời gian giữa thời điểm xảy ra biến cố với thời điểm sử dụng thuốc. Một số phản ứng có thể xảy ra ngay lập tức sau khi sử dụng thuốc, trong khi cũng có những phản ứng diễn biến chậm hơn và sau một thời gian mới xuất hiện.
– Thăm khám bệnh nhân thường xuyên và tiến hành các xét nghiệm cần thiết. Kết quả xét nghiệm rất có ích trong việc phát hiện sớm bất thường cận lâm sàng và có thể được sử dụng để đo lường mức độ nghiêm trọng cũng như theo dõi giám sát người bệnh.
– Ngừng thuốc và kiểm tra lại thông tin tác dụng dược lý của thuốc và vị thuốc.
– Kiểm tra phản ứng xảy ra đã được liệt kê ở các tài liệu tra cứu về thuốc hay chưa (tờ hướng dẫn sử dụng thuốc, bản tóm tắt đặc tính sản phẩm trong hồ sơ đăng ký thuốc, các tài liệu tham khảo tin cậy khác). Nếu phản ứng chưa được biết đến và không giải thích được bằng cơ chế tác dụng dược lý thì nên lưu ý trong quá trình theo dõi, xử lý và báo cáo.
3.2. Giám sát chất lượng thuốc y học cổ truyền
Kết quả kiểm nghiệm chất lượng dược liệu, thuốc đông y, thuốc từ dược liệu của các cơ quan kiểm định chất lượng thuốc (bao gồm Viện kiểm nghiệm thuốc Trung ương, Viện kiểm nghiệm thuốc thành phố Hồ Chí Minh và các Trung tâm kiểm nghiệm các tỉnh/ thành phố) hoặc Hội đồng kiểm nhập trong các cơ sở khám bệnh, chữa bệnh cần được báo cáo cho cơ quan quản lý để kịp thời phát hiện thuốc YHCT không đảm bảo chất lượng. Thông qua kết quả kiểm nghiệm, cơ quan quản lý sẽ khuyến cáo về chất lượng dược liệu, vị thuốc y học cổ truyền trên toàn bộ hệ thống quản lý y dược học cổ truyền.
3.2.1. Phát hiện thuốc y học cổ truyền không đạt tiêu chuẩn chất lượng
a) Đối với dược liệu
– Dược liệu không đạt tiêu chuẩn chủ yếu không đạt về độ ẩm, tạp chất, định tính, hàm lượng hoạt chất (ví dụ Hà thủ ô đỏ không có hoặc có rất ít emodin, Hoàng cầm không đạt hàm lượng baicalin, Cam thảo không đạt hàm lượng acid glycyrrhizic, …).
– Dược liệu có chứa các chất nguy hại: acid aristolochic (tìm thấy trong Phòng kỷ, Mộc thông, Tế tân, …), β-asaron (tìm thấy trong Thạch xương bồ), aflatoxin, dư lượng thuốc bảo vệ thực vật, kim loại nặng. Một số dược liệu được nhuộm màu, tẩm hóa chất để làm “đẹp” như Hồng hoa, Chi tử nhuộm rhodamin B, …
– Dược liệu giả: là các dược liệu có hình thái mô tả gần giống với dược liệu thật nhưng không có các đặc điểm thực vật và hoá học giống với dược liệu thật, không đúng bộ phận dùng hay bị trộn lẫn dược liệu thật với thành phần khác theo các tỷ lệ khác nhau nhưng vẫn lấy tên là dược liệu thật. Ví dụ:
+ Hoàng kỳ đang lưu hành trên thị trường có thể khác với dược liệu Hoàng kỳ [Astragalusmembranaceus (Fisch.) Bge., Fabaceae] (theo tiêu chuẩn Dược điển Việt Nam (DĐVN) IV và Dược điển Trung Quốc) và giống dược liệu Hồng kỳ [Hedsarum polybotrys Hand., Mazz, Fabaceae] đã được ghi trong Dược điển Trung Quốc; khi định tính và định lượng không có hoạt chất astragalosid IV.
+ Hoài sơn: lẫn với các củ khác thuộc họ Dioscoreaceae như Củ mỡ. + Đinh lăng: dùng thân, cành để thay thế rễ.
+ Thăng ma: dùng rễ của cây khác có hình thái gần giống nhưng không có các hoạt chất của Thăng ma như acid ferulic và acid isoferulic.
– Dược liệu trộn các chất khác: trộn các chất làm tăng màu, tăng khối lượng: Hồng hoa và Chi tử trộn rhodamin B làm tăng màu, Bạch linh làm giả hoàn toàn hay trộn lẫn với các loại bột khác, ép thành bánh giống Bạch linh, Thỏ ty tử trộn xi măng hoặc loại hạt khác, …
– Dược liệu chế biến chưa đúng quy định có thể gây tác dụng có hại. Ví dụ Viễn chí không bỏ hết lõi có thể gây nôn và buồn nôn, Ô đầu không chế hoặc chế không đúng quy định gây tụt huyết áp hoặc trụy tim mạch, …
– Dược liệu mốc mọt hoặc dùng thuốc bảo quản thực vật không cho phép.
b) Đối với thuốc đông y, thuốc từ dược liệu
– Thuốc đông y, thuốc từ dược liệu chủ yếu không đạt những tiêu chuẩn sau: giới hạn nhiễm khuẩn (số mẫu hàng năm không đạt yêu cầu về giới hạn nhiễm khuẩn chiếm tỷ lệ cao trong số các chỉ tiêu không đạt); độ ẩm; định tính, định lượng hoạt chất; các chỉ tiêu về kỹ thuật bào chế như độ rã, đồng đều khối lượng.
– Thuốc đông y, thuốc từ dược liệu giả: việc trộn các thuốc tân dược vào thuốc cổ truyền rất khó phát hiện, ngay cả khi đem kiểm nghiệm, vì trong thuốc có rất nhiều thành phần, đòi hỏi phương tiện kiểm nghiệm hiện đại, tốn nhiều thời gian và chi phí.
– Thuốc trộn trái phép tân dược (thành phần có hoạt chất tân dược nhưng không công bố trên nhãn), các hoạt chất tân dược được dùng để trộn lẫn bao gồm nhiều nhóm hợp chất khác nhau, trong đó đáng chú ý nhất là:
+ Nhóm hoạt chất tăng cường chức năng sinh dục (sildenafil và dẫn chất).
+ Nhóm corticosteroid (dexamethason, prednisolon, betamethason, … ).
+ Nhóm thuốc kháng histamin (clopheniramin, cyproheptadin, … ).
+ Nhóm thuốc chống viêm không steroid (diclofenac, ibuprofen, indomethacin, … ).
+ Nhóm thuốc hạ sốt giảm đau (paracetamol, aspirin, … ). + Nhóm thuốc an thần gây ngủ (diazepam, zolpidem, … ). + Nhóm thuốc giảm béo (sibutramin, dimethylamylamin, …).
Thuốc tân dược trộn vào thuốc YHCT hoặc thực phẩm chức năng hay gặp trong điều trị thấp khớp, đái tháo đường, suy giảm chức năng sinh dục nam, viêm xoang, thừa cân, béo phì có thể gây ra nhiều hậu quả nghiêm trọng. Hình thức giả mạo thường gặp là trộn thuốc tân dược dạng bột hoặc được tán thành bột vào bột dược liệu rồi chế thành thành phẩm ở dạng viên, dạng cao lỏng hay dạng thuốc sắc. Sự việc đáng chú ý là việc trộn chì vào thuốc cam gây ngộ độc chì cho trẻ em. Kết quả kiểm nghiệm, đã phát hiện một số loại thuốc đó có hàm lượng chì (Pb) cao (từ 0,1% đến 3,5%).
3.2.2. Một số yếu tố cần xem xét khi phân tích các vấn đề liên quan đến đảm bảo chất lượng thuốc y học cổ truyền
Vấn đề về chất lượng thuốc y học cổ truyền có thể do nhiều nguyên nhân bao gồm:
– Nguồn gốc: nguồn dược liệu sử dụng trong nước chủ yếu được nhập khẩu từ Trung Quốc theo con đường tiểu ngạch hoặc sản xuất nhỏ lẻ trong nhân dân, chưa có nhiều vùng nuôi trồng dược liệu lớn được kiểm soát chặt chẽ đạt tiêu chuẩn Thực hành tốt trồng trọt và thu hái cây thuốc theo hướng dẫn của Tổ chức Y tế Thế giới (GACP-WHO). Do vậy, việc kiểm soát được nguồn gốc cũng như chất lượng dược liệu gặp nhiều khó khăn, dẫn đến tình trạng dược liệu kém, nhầm lẫn, thay thế, giả mạo vẫn còn phổ biến trên thị trường. Dược liệu được sử dụng làm thuốc và nguyên liệu sản xuất đông dược rất khó kiểm soát nguồn gốc và truy tìm do không có số lô, hạn dùng. Điều này cũng gây khó khăn trong việc thu hồi xử lý các mẫu không đạt chất lượng hoặc bị làm giả.
– Sản xuất và chế biến: một số chế phẩm YHCT sản xuất trong các cơ sở chưa đạt tiêu chuẩn Thực hành sản xuất thuốc tốt (GMP), không được kiểm soát chặt chẽ về quy trình sản xuất cũng như kiểm soát về chất lượng nguyên liệu đầu vào và sản phẩm xuất xưởng. Quy trình chế biến, bào chế dược liệu không tuân thủ theo quy định của Bộ Y tế, chưa tiêu chuẩn hóa các vị thuốc sau khi chế biến, nhất là các vị thuốc có độc tính, dẫn đến chất lượng dược liệu sau chế biến không đảm bảo. Thiếu kiểm tra hoặc kiểm tra không đầy đủ nguyên liệu đầu vào dẫn đến sử dụng dược liệu kém chất lượng hoặc dược liệu không đúng để sản xuất. Nhân lực còn thiếu, yếu và chưa được đào tạo hoặc đào tạo chưa đầy đủ về kiến thức chuyên môn.
– Tiêu chuẩn: các tiêu chuẩn thuốc từ dược liệu và dược liệu còn chưa đề cập nhiều về vấn đề định lượng hoạt chất mà mới chủ yếu dừng ở mức định tính. Hiện DĐVN IV có 314 chuyên luận về dược liệu và chế phẩm YHCT. So với DĐVN III, một số các chuyên luận này đã được bổ sung chỉ tiêu về định tính, định lượng hoạt chất và độ an toàn (ví dụ như kim loại nặng) tuy nhiên vẫn còn nhiều chuyên luận chưa có các chỉ tiêu này và chưa có quy định cụ thể trong chuyên luận về kiểm tra aflatoxin, dư lượng hóa chất bảo vệ thực vật, …
– Chất chuẩn: hiện rất thiếu chất chuẩn, dược liệu chuẩn dùng trong kiểm tra chất lượng dược liệu, đặc biệt trong kiểm tra chất lượng dược liệu có nguồn gốc nhập khẩu. Việc sử dụng các chất chuẩn cũng như dược liệu chuẩn trong kiểm tra chất lượng đã được nghiên cứu và thực hiện nhưng chưa được hệ thống hóa và kết nối với nhau.
– Công tác đấu thầu: thuốc nhập khẩu qua đường tiểu ngạch có giá thấp hơn nhiều so với giá trị vốn có của chúng. Trong công tác đấu thầu lựa chọn thuốc đưa vào cơ sở khám bệnh, chữa bệnh, những thuốc có giá thành thấp sẽ trúng thầu. Vì vậy, người kinh doanh có thể vì lợi nhuận thương mại đưa hàng kém chất lượng vào bệnh viện. Tại cơ sở khám bệnh, chữa bệnh YHCT tư nhân, các thầy thuốc có thể không đủ trình độ, kinh nghiệm để nhận biết thuốc kém chất lượng hoặc có xu hướng sử dụng các thuốc YHCT giá rẻ mà không đặt cao yêu cầu chất lượng thuốc.
– Bảo quản: điều kiện bảo quản dược liệu chưa đảm bảo nên dễ làm giảm chất lượng của thuốc y học cổ truyền. Nhiều loại dược liệu dễ bị ẩm mốc nhưng không được xử lý trước khi đưa vào sử dụng nên không đảm bảo an toàn. Ngược lại, việc dùng diêm sinh quá mức trong bảo quản dược liệu khá phổ biến, đặc biệt là những dược liệu loại củ chứa nhiều tinh bột bắt buộc phải qua công đoạn sấy diêm sinh để chống mốc, chống thối khi đưa vào sử dụng.
3.2.3. Xử lý các vấn đề liên quan đến chất lượng thuốc
– Tăng cường công tác kiểm tra giám sát chất lượng thuốc YHCT. Xử lý nghiêm các trường hợp cố ý trộn trái phép tân dược vào thuốc YHCT.
– Nguốc gốc: xây dựng, phát triển nguồn dược liệu sạch, sử dụng các loại dược liệu có nguồn gốc xuất xứ rõ ràng. Cần sớm triển khai GACP-WHO với các dược liệu mà Việt Nam có thế mạnh phát triển.
– Kiểm nhập: tăng cường giám sát chất lượng thuốc ngay từ khâu nhập vào cơ sở khám bệnh, chữa bệnh, đảm bảo việc nhập thuốc đúng và đạt chất lượng.
– Sản xuất và chế biến: đảm bảo vệ sinh và tuân thủ việc chế biến theo đúng các quy định của Bộ Y tế. Nếu các vị thuốc có độc phải chế biến tuân thủ đúng quy định của Bộ Y tế và có phiếu kiểm nghiệm sau chế biến và việc đưa vào sử dụng trong các đơn vị bắt buộc phải có phiếu kiểm nghiệm. Đảm bảo thực hiện đúng lộ trình GMP đối với cơ sở sản xuất thuốc từ dược liệu, đồng thời có chế độ chính sách khuyến khích các đơn vị thực hiện sớm. Khảo sát, nghiên cứu và cập nhật kết quả nghiên cứu hoặc thông báo về các chất có độc tính có trong dược liệu.
– Bảo quản: dược liệu, vị thuốc y học cổ truyền cần được bảo quản theo nguyên tắc tiêu chuẩn Thực hành bảo quản thuốc tốt (GSP). Chỉ các đơn vị có chứng nhận GSP mới được kinh doanh và buôn bán dược liệu, thuốc YHCT. Các kho chứa dược liệu phải được thiết kế đúng tiêu chuẩn. Dược liệu, vị thuốc YHCT phải được dán nhãn đầy đủ, tránh nhầm lẫn. Dược liệu, vị thuốc YHCT phải được kiểm tra, kiểm soát, kiểm nghiệm chất bảo quản, dư lượng chất bảo vệ thực vật. Thuốc từ dược liệu, thuốc đông y bắt buộc phải có giới hạn chất bảo quản.
– Tăng cường nguồn chất chuẩn và dược liệu chuẩn dùng trong công tác kiểm tra giám sát chất lượng dược liệu. Tiếp tục bổ sung các chỉ tiêu về định tính, định lượng hoạt chất, các chỉ tiêu về độ an toàn (dư lượng thuốc bảo vệ thực vật, aflatoxin, kim loại nặng) trong tiêu chuẩn đánh giá chất lượng dược liệu và thuốc từ dược liệu.
– Truyền thông: tuyên truyền sử dụng đông dược hợp lý, tránh để người dân sử dụng thuốc YHCT không rõ nguồn gốc xuất xứ gây nguy hiểm đến tính mạng.
– Sử dụng: cân và cấp phát theo đúng đơn thuốc được kê. Đặc biệt chú trọng việc đảm bảo độ đồng đều khi chia vị thuốc y học cổ truyền vào các thang thuốc.
3.3. Sai sót liên quan đến sử dụng thuốc y học cổ truyền
3.3.1. Các hình thức sai sót liên quan đến thuốc y học cổ truyền
a) Sai sót trong quá trình kê đơn
– Phối ngũ sai trong quá trình kê đơn: hai vị thuốc tương tác trong cùng một đơn thuốc làm tăng độc tính của thuốc.
– Không ghi rõ cách chế biến vị thuốc YHCT làm ảnh hưởng tới tác dụng của thuốc và không đảm bảo an toàn cho người sử dụng. Ví dụ: Chích Hoàng kỳ (Hoàng kỳ chích mật) không dùng cho người bị đái tháo đường mà dùng Sinh Hoàng kỳ (Hoàng kỳ sống); Phụ tử (chưa chế biến) có độc tính cao còn việc sử dụng Hắc phụ (Phụ tử đã chế biến) sẽ giúp làm giảm độc tính của dược liệu.
– Không ghi rõ hướng dẫn cách sắc thuốc trong đơn thuốc. Ví dụ: không ghi rõ khi sắc Ma hoàng phải hớt bỏ bọt.
– Không ghi rõ thời điểm uống thuốc (lúc đói, lúc no, …).
– Không ghi kiêng kỵ khi dùng thuốc. Ví dụ: không ghi rõ khi đang dị ứng thì kiêng thức ăn tanh.
b) Sai sót trong quá trình bào chế
Bào chế là một quá trình chế biến dược liệu mang tính độc đáo của dược học cổ truyền nhằm tăng tác dụng của thuốc hoặc giảm bớt độc tính của thuốc. Nếu quá trình bào chế không được thực hiện đúng quy định sẽ không loại trừ được độc tính của thuốc và có thể gây phản ứng có hại khi sử dụng.
Trong YHCT có nhiều vị thuốc dễ gây ngộ độc, nôn mửa nếu bào chế không kỹ như bán hạ chế, phụ tử chế, … Vị thuốc tỳ bà diệp (lá nhót) khi bào chế phải làm sạch các lông tơ trên mặt lá để tránh kích ứng niêm mạc họng, gây ho, sưng niêm mạc.
c) Sai sót trong quá trình sắc thuốc, cấp phát, hướng dẫn sử dụng
Nhầm lẫn khi sắc thuốc có thể xảy ra nếu không đánh số thang thuốc, phiếu, ấm sắc thuốc, bình đựng thuốc trước khi sắc thuốc; không có tủ giá để sắp xếp phân biệt thuốc chưa sắc, thuốc đang sắc dở, thuốc đã sắc xong; hệ thống sổ sách theo dõi quá trình sắc thuốc và cấp phát thuốc sắc chưa được ghi chép đầy đủ, chưa theo biểu mẫu quy định.
d) Sai sót trong cách dùng thuốc
Nhiều vị thuốc độc tính cao thường chỉ dùng bôi, đắp ngoài da. Khi dùng đường uống có thể gây những tác hại nặng nề, có khi dẫn tới tử vong. Ví dụ mật cá trắm, lá vòi voi dùng đắp ngoài chữa các bệnh khớp, … lại dùng đường uống dẫn đến suy thận cấp, hoại tử ống thận cấp rất nguy hiểm.
e) Sai sót trong quá trình người bệnh sử dụng thuốc
Việc người bệnh không tuân thủ về cách dùng, thời điểm dùng thuốc theo lời dặn của thầy thuốc, tự ý dùng thuốc YHCT không rõ nguồn gốc hoặc qua truyền miệng có thể là những sai sót dẫn đến xuất hiện phản ứng có hại hoặc độc tính của thuốc YHCT.
3.3.2. Cách hạn chế sai sót
– Bào chế và quản lý thuốc sắc phải do dược sĩ hoặc nhân viên y tế được đào tạo tại cơ sở khám bệnh, chữa bệnh phụ trách.
– Chống nhầm lẫn khi tiến hành sắc thuốc: đánh số vào thang thuốc, phiếu, ấm sắc thuốc, bình đựng thuốc trước và sau khi sắc thuốc; phải có tủ giá để sắp xếp phân biệt thuốc chưa sắc, thuốc đang sắc dở, thuốc đã sắc xong.
– Tổ chức phát thuốc hàng ngày và thuốc bổ sung theo y lệnh.
– Có sổ xuất nhập hằng ngày để ghi chép số thang thuốc đã nhận, đã giao và số còn lại trong ngày.
– Với những bài thuốc có chứa dược liệu, vị thuốc y học cổ truyền có độc tính: cần lưu riêng bã thuốc khi đã sắc xong.
3.4. Báo cáo phản ứng có hại của thuốc và các vấn đề liên quan đến thuốc
– Các trường hợp bắt buộc phải báo cáo: khi có biểu hiện ngộ độc cấp tính, ngộ độc mạn tính nghiêm trọng do sử dụng dược liệu, vị thuốc y học cổ truyền, thuốc đông y, thuốc từ dược liệu.
– Thời gian gửi báo cáo, hình thức gửi báo cáo và nơi nhận báo cáo: theo hướng dẫn tại chương 2 (mục 2.1.5).
– Biểu mẫu báo cáo: tham khảo mẫu tại phụ lục 10 của Hướng dẫn này. Mẫu báo cáo này tương tự mẫu báo cáo chung về phản ứng có hại của thuốc, trong đó lưu ý với thuốc YHCT nghi ngờ gây ra phản ứng, cần ghi rõ tên dược liệu, vị thuốc đông y, khuyến khích ghi thêm tên khoa học của vị thuốc, ngày chế biến và ngày sử dụng thuốc đó.
CHƯƠNG 4.
HOẠT ĐỘNG CẢNH GIÁC DƯỢC
TRONG CHƯƠNG TRÌNH TIÊM CHỦNG
Trong chương trình tiêm chủng, hoạt động Cảnh giác dược chủ yếu đang được triển khai là giám sát phản ứng sau tiêm chủng (PƯSTC). Giám sát PƯSTC bao gồm theo dõi, phát hiện sớm, xử trí và báo cáo các PƯSTC để giảm bớt tác động không tốt đến sức khỏe của người được tiêm chủng và cung cấp số liệu thực tế về tính an toàn của vắc xin và thực hành tiêm chủng. Chương này sẽ tổng hợp các nội dung cần lưu ý đối với nhân viên y tế tại các cơ sở có hoạt động tiêm chủng trong công tác giám sát PƯSTC được Bộ Y tế quy định, hướng dẫn theo Thông tư số 12/2014/TT-BYT ngày 20 tháng 03 năm 2014 về việc hướng dẫn việc quản lý sử dụng vắc xin trong tiêm chủng và Quyết định số 1830/QĐ-BYT ban hành ngày 26 tháng 05 năm 2014 về việc hướng dẫn giám sát, điều tra, phân tích, đánh giá nguyên nhân phản ứng sau tiêm chủng.
4.1. Định nghĩa, phân loại phản ứng sau tiêm chủng
4.1.1. Định nghĩa
Phản ứng sau tiêm chủng là hiện tượng bất thường về sức khỏe bao gồm các biểu hiện tại chỗ tiêm chủng hoặc toàn thân xảy ra sau tiêm chủng. PƯSTC bao gồm phản ứng thông thường sau tiêm chủng và tai biến nặng sau tiêm chủng.
4.1.2. Phân loại
a) Theo mức độ
– Phản ứng thông thường sau tiêm chủng bao gồm các phản ứng tại chỗ như ngứa, đau, sưng và/hoặc đỏ tại chỗ tiêm; phản ứng toàn thân bao gồm sốt và các triệu chứng khác (khó chịu, mệt mỏi, chán ăn), có thể là một phần của đáp ứng miễn dịch bình thường. Các phản ứng này thông thường là nhẹ và tự khỏi.
– Tai biến nặng sau tiêm chủng là phản ứng bất thường sau tiêm chủng có thể đe dọa đến tính mạng người được tiêm chủng (bao gồm các triệu chứng như khó thở, sốc phản vệ hay sốc dạng phản vệ, hội chứng sốc nhiễm độc, sốt cao co giật, trẻ khóc kéo dài, tím tái, ngừng thở) hoặc để lại di chứng hoặc làm người được tiêm chủng tử vong.
b) Theo nguyên nhân
– Do trùng hợp ngẫu nhiên: xảy ra sau khi tiêm chủng nhưng nguyên nhân không phải do vắc xin hoặc sai sót trong tiêm chủng hoặc lo sợ do bị tiêm mà do trùng hợp ngẫu nhiên với bệnh lý sẵn có hoặc nguyên nhân khác.
– Do tâm lý lo sợ: xảy ra do sự lo sợ hoặc do bị tiêm đau, không phải do vắc xin hoặc sai sót trong thực hành tiêm chủng.
– Do vắc xin: phản ứng sau tiêm chủng xảy ra do các đặc tính cố hữu của vắc xin hoặc do vắc xin không đạt chất lượng.
– Do sai sót trong thực hành tiêm chủng: xảy ra do sai sót trong quá trình thực hành tiêm chủng (chuẩn bị, pha hồi chỉnh, kỹ thuật tiêm, bảo quản và sử dụng vắc xin không đúng).
– Không rõ nguyên nhân: không xác định được nguyên nhân.
4.2. Hướng dẫn giám sát phản ứng sau tiêm chủng
4.2.1. Sơ đồ hệ thống giám sát
Sơ đồ hệ thống giám sát phản ứng sau tiêm chủng được thể hiện trong hình 3.
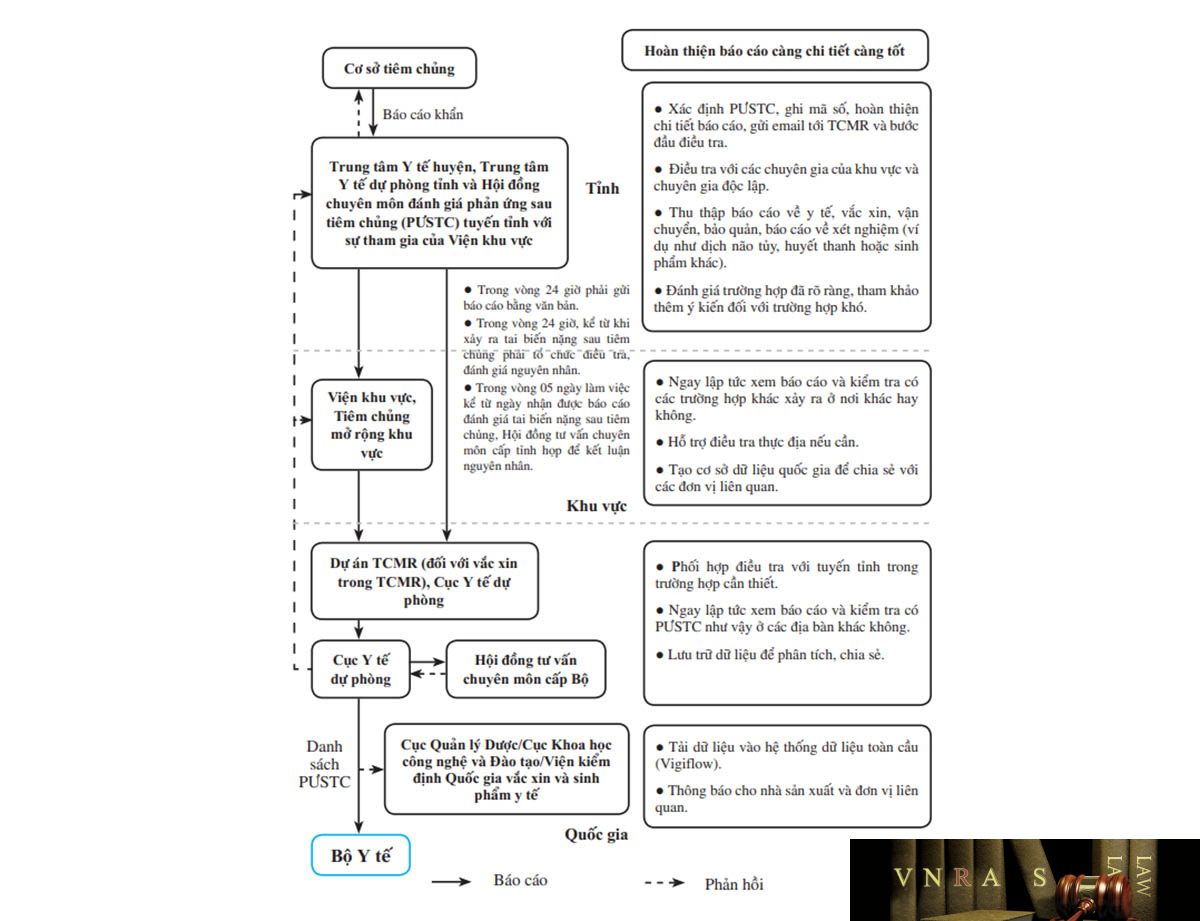
Hình 3. Hệ thống báo cáo tai biến nặng sau tiêm chủng
4.2.2. Phát hiện, xử trí tai biến nặng sau tiêm chủng
a) Tất cả đối tượng sau khi tiêm chủng phải được theo dõi tối thiểu 30 phút tại địa điểm tiêm chủng, sau đó tiếp tục theo dõi tại nhà ít nhất 24 giờ sau khi tiêm chủng để phát hiện sớm những tai biến nặng sau tiêm chủng.
b) Tại cơ sở tiêm chủng: khi đang triển khai tiêm chủng mà xảy ra tai biến nặng sau tiêm chủng, người đứng đầu cơ sở tiêm chủng phải chỉ đạo thực hiện các nội dung sau đây:
– Dừng ngay buổi tiêm chủng;
– Xử trí cấp cứu, chẩn đoán nguyên nhân tai biến nặng sau tiêm chủng; trường hợp vượt quá khả năng thì phải chuyển người bị tai biến nặng sau tiêm chủng đến bệnh viện gần nhất;
– Ghi chép đầy đủ thông tin:
+ Họ tên, tuổi, giới, địa chỉ, số điện thoại (nếu có) của người được tiêm. Họ tên bố, mẹ, địa chỉ, số điện thoại (nếu có) đối với trẻ em;
+ Ngày, giờ tiêm chủng;
+ Loại vắc xin; tên vắc xin; số lô; hạn dùng; nhà sản xuất; đơn vị cung cấp vắc xin; số đăng ký lưu hành hoặc số giấy phép nhập khẩu;
+ Ngày, giờ xuất hiện tai biến nặng sau tiêm chủng; các triệu chứng chính; kết quả điều trị; kết luận nguyên nhân (nếu có);
– Thống kê toàn bộ số lượng vắc xin đã sử dụng trong buổi tiêm chủng; số người đã được sử dụng theo loại vắc xin; tên vắc xin, số lô, hạn dùng của vắc xin; tình trạng sức khỏe của những người đã được tiêm chủng;
– Thống kê toàn bộ số vỏ lọ vắc xin, bơm kim tiêm đã sử dụng trong buổi tiêm chủng;
– Báo cáo tuyến trên (chi tiết xem phần 4.2.3 của hướng dẫn này).
c) Tại nơi tiếp nhận trường hợp tai biến nặng sau tiêm chủng, người đứng đầu nơi tiếp nhận phải chỉ đạo thực hiện các nội dung sau:
– Tiến hành cấp cứu, xử trí và điều trị theo quy định;
– Báo cáo tuyến trên (chi tiết xem phần 4.2.3 của hướng dẫn này).
d) Tuyến huyện, tỉnh: tiếp nhận báo cáo, tổng hợp thông tin, báo cáo tuyến trên (chi tiết xem phần 4.2.3 của hướng dẫn này).
4.2.3. Chế độ báo cáo và quản lý hồ sơ trường hợp phản ứng sau tiêm chủng
a) Chế độ báo cáo
– Báo cáo định kỳ: báo cáo tháng, quý và năm về các trường hợp phản ứng thông thường và các trường hợp tai biến nặng sau tiêm chủng;
– Báo cáo đột xuất: các trường hợp tai biến nặng sau tiêm chủng.
b) Hình thức, nội dung báo cáo
Hình thức báo cáo:
– Báo cáo định kỳ: bằng văn bản;
– Báo cáo đột xuất: trong trường hợp khẩn cấp có thể báo cáo qua điện thoại, thư điện tử, hoặc báo cáo trực tiếp và trong vòng 24 giờ phải gửi báo cáo bằng văn bản.
Nội dung báo cáo:
– Báo cáo định kỳ:
+ Báo cáo các trường hợp phản ứng thông thường sau tiêm chủng theo quy định hiện hành (xem phụ lục 11 của Hướng dẫn này);
+ Báo cáo các trường hợp tai biến nặng sau tiêm chủng theo quy định hiện hành (xem phụ lục 12 của Hướng dẫn này).
– Báo cáo đột xuất: trường hợp tai biến nặng sau tiêm chủng theo quy định hiện hành (xem phụ lục 13 của Hướng dẫn này) hoặc theo yêu cầu của cơ quan đề nghị báo cáo.
c) Quy trình và thời gian báo cáo định kỳ
Đối với vắc xin trong Chương trình Tiêm chủng mở rộng
– Cơ sở tiêm chủng: báo cáo Trung tâm Y tế huyện trước ngày 05 của tháng tiếp theo đối với báo cáo tháng, ngày 05 của tháng đầu tiên của quý tiếp theo đối với báo cáo quý, trước ngày 15 tháng 01 của năm tiếp theo đối với báo cáo năm;
– Trung tâm y tế huyện: báo cáo Trung tâm Y tế dự phòng tỉnh trước ngày 10 của tháng tiếp theo đối với báo cáo tháng, ngày 10 của tháng đầu tiên của quý tiếp theo đối với báo cáo quý, trước ngày 25 tháng 01 của năm tiếp theo đối với báo cáo năm;
– Trung tâm Y tế dự phòng tỉnh: báo cáo Sở Y tế, Dự án Tiêm chủng mở rộng khu vực tại các Viện Vệ sinh dịch tễ trung ương, Viện Pasteur thành phố Hồ Chí Minh, Viện Pasteur Nha Trang, Viện Vệ sinh tễ trung ương (sau đây gọi tắt là Viện khu vực) theo phân vùng quản lý của Bộ trưởng Bộ Y tế, đồng thời báo cáo Dự án Tiêm chủng mở rộng quốc gia trước ngày 15 của tháng tiếp theo đối với báo cáo tháng, ngày 15 của tháng đầu tiên của quý tiếp theo đối với báo cáo quý, trước ngày 31 tháng 01 của năm tiếp theo đối với báo cáo năm;
– Dự án Tiêm chủng mở rộng quốc gia tổng hợp báo cáo, gửi Cục Y tế dự phòng (Bộ Y tế) trước ngày 20 của tháng tiếp theo đối với báo cáo tháng, ngày 20 của tháng đầu tiên của quý tiếp theo đối với báo cáo quý, trước ngày 15 tháng 02 của năm tiếp theo đối với báo cáo năm.
Đối với vắc xin tiêm chủng dịch vụ
– Cơ sở tiêm chủng: báo cáo Trung tâm Y tế huyện trước ngày 05 của tháng tiếp theo đối với báo cáo tháng, ngày 05 của tháng đầu tiên của quý tiếp theo đối với báo cáo quý, trước ngày 15 tháng 01 của năm tiếp theo đối với báo cáo năm;
– Trung tâm Y tế huyện: báo cáo Trung tâm Y tế dự phòng tỉnh trước ngày 10 của tháng tiếp theo đối với báo cáo tháng, ngày 10 của tháng đầu tiên của quý tiếp theo đối với báo cáo quý, trước ngày 25 tháng 01 của năm tiếp theo đối với báo cáo năm;
– Trung tâm Y tế dự phòng tỉnh: báo cáo Sở Y tế, các Viện khu vực, Cục Y tế dự phòng, Bộ Y tế trước ngày 15 của tháng tiếp theo đối với báo cáo tháng, ngày 15 của tháng đầu tiên của quý tiếp theo đối với báo cáo quý, trước ngày 31 tháng 01 của năm tiếp theo đối với báo cáo năm.
d) Quy trình và thời gian báo cáo đột xuất
– Trong thời gian 24 giờ, kể từ thời điểm ghi nhận tai biến nặng sau tiêm chủng, cơ sở tiêm chủng báo cáo Trung tâm Y tế dự phòng tỉnh, đồng thời báo cáo Trung tâm Y tế huyện nơi cơ sở tiêm chủng đặt trụ sở.
– Trong thời gian 24 giờ, kể từ thời điểm nhận được báo cáo của cơ sở tiêm chủng, Trung tâm Y tế dự phòng tỉnh báo cáo Sở Y tế, Cục Y tế dự phòng và Viện khu vực.
– Hàng tuần, sau khi thực hiện việc báo cáo quy định ở trên:
+ Cơ sở tiêm chủng phải thực hiện báo cáo diễn biến quá trình điều tra, xử lý trong tuần vào ngày thứ 2 của tuần kế tiếp;
+ Trung tâm Y tế dự phòng tỉnh phải thực hiện báo cáo diễn biến quá trình điều tra, xử lý trong tuần vào ngày thứ 4 của tuần kế tiếp.
4.3. Điều tra tai biến nặng sau tiêm chủng
4.3.1. Thành phần đoàn điều tra
Theo quy định tại Thông tư số 21/2011/TT-BYT ban hành ngày 07 tháng 06 năm 2011 của Bộ trưởng Bộ Y tế quy định việc thành lập, tổ chức và hoạt động của Hội đồng tư vấn chuyên môn đánh giá tai biến trong quá trình sử dụng vắc xin, sinh phẩm y tế.
4.3.2. Quy trình điều tra
– Sử dụng phiếu điều tra tai biến nặng sau tiêm chủng theo hướng dẫn hiện hành (xem phụ lục 14 của Hướng dẫn này).
– Điền đầy đủ các thông tin trong phiếu điều tra.
– Điền mã số trường hợp tai biến nặng sau tiêm bao gồm chữ viết tắt của tỉnh, huyện và điểm tiêm chủng, cuối cùng là số thứ tự trường hợp tai biến tại điểm tiêm chủng đó được viết dưới dạng số, ví dụ trường hợp thứ nhất tai biến nặng sau tiêm chủng tại xã A huyện B, tỉnh C: mã số được viết như sau: VNCBA01.
– Các bước điều tra cụ thể được trình bày trong bảng 2 như sau:
Bảng 2. Quy trình điều tra tai biến nặng sau tiêm chủng
| TT | Các bước | Hành động |
| 1 | Xác minh các thông tin trong báo cáo | – Thu thập hồ sơ bệnh án (hoặc ghi chép về lâm sàng). – Kiểm tra hồ sơ chi tiết về người bệnh, tình trạng diễn biến sức khỏe. – Phỏng vấn nhân viên y tế tiếp nhận, điều trị trường hợp tai biến nặng sau tiêm chủng, rà soát hồ sơ bệnh án. – Thu thập thêm thông tin còn thiếu trong báo cáo. – Xác định những trường hợp khác cần điều tra. |
| 2 | Điều tra tai biến nặng sau tiêm chủng | |
| 2.1. Điều tra và thu thập thông tin từ người bệnh hoặc người nhà | – Tiền sử tiêm chủng. – Tiền sử bệnh tật, bao gồm tiền sử về phản ứng tương tự hoặc những tình trạng dị ứng khác. – Tiền sử về gia đình đối với những phản ứng tương tự. – Phỏng vấn trực tiếp bà mẹ hoặc người chăm sóc trẻ, rà soát hồ sơ liên quan tới trường hợp phản ứng sau tiêm chủng do người nhà giữ. |
|
| 2.2. Điều tra trường hợp tai biến nặng sau tiêm chủng | – Bệnh sử, mô tả lâm sàng, tất cả các xét nghiệm liên quan đến tai biến nặng sau tiêm chủng và chẩn đoán. – Điều trị, kết quả. |
|
| 2.3. Điều tra vắc xin nghi ngờ | – Điều kiện vận chuyển vắc xin, điều kiện bảo quản hiện tại, tình trạng bảo quản lọ vắc xin, bảng theo dõi nhiệt độ tủ lạnh. – Bảo quản vắc xin trước khi đến cơ sở y tế, phiếu tiếp nhận vắc xin, thẻ chỉ thị nhiệt. |
|
| 2.4. Điều tra hững người liên quan | – Những người đã được tiêm chủng cùng loại vắc xin trong cùng một buổi tiêm chủng có phản ứng hay không? phản ứng tương tự hay không? – Điều tra dịch vụ tiêm chủng. |
|
| 3 | Đánh giá thực hành tiêm chủng | |
| 3.1. Đánh giá dịch vụ bằng cách hỏi | – Đánh giá điểm tiêm chủng: hỏi, quan sát việc cung cấp dịch vụ tiêm chủng, bảo quản vắc xin. – Cách bảo quản vắc xin (kể cả những lọ đã mở), việc phân phối và hủy bỏ vắc xin. – Cách bảo quản và phân phối dung môi. – Việc pha hồi chỉnh vắc xin (kỹ thuật và thời gian sử dụng sau khi pha). – Cách sử dụng và vô trùng bơm, kim tiêm. – Những chi tiết về huấn luyện thực hành tiêm chủng, về giám sát các kỹ thuật tiêm chủng. |
|
| 3.2. Quan sát dịch vụ tiêm chủng | – Tủ lạnh: ngoài vắc xin còn bảo quản thêm những gì trong tủ lạnh (cần ghi chép nếu có những lọ tương tự được để cạnh những lọ vắc xin có thể nhầm lẫn); những loại vắc xin hoặc dung môi nào để cùng với những loại thuốc khác; có lọ vắc xin, sinh phẩm y tế nào mất nhãn, quá hạn sử dụng không? – Thực hành tiêm chủng (hồi chỉnh vắc xin, mở nút lọ, kỹ thuật tiêm, đảm bảo an toàn bơm, kim tiêm, vứt bỏ những lọ đã mở). |
|
| 4 | Đặt giả thuyết | – Nguyên nhân có thể xảy ra. |
| 5 | Kiểm tra giả thuyết | – Trường hợp phản ứng có phù hợp với giả thuyết? – Chỉ lấy mẫu và yêu cầu kiểm định vắc xin nếu nghi ngờ nguyên nhân do vắc xin. |
| 6 | Kết thúc điều tra | – Hoàn chỉnh phiếu điều tra. – Kết luận của đoàn điều tra. – Khuyến nghị. |
4.3.3. Lấy mẫu vắc xin để kiểm định
– Lấy mẫu và yêu cầu kiểm định vắc xin: tùy thuộc vào giả thuyết về nguyên nhân của tai biến sau tiêm chủng. Chỉ lấy mẫu vắc xin khi nghi ngờ nguyên nhân do vắc xin. Trung tâm Y tế dự phòng tỉnh, thành phố lấy mẫu vắc xin, ghi vào Phiếu lấy và gửi mẫu kiểm định vắc xin theo quy định hiện hành (xem phụ lục 15 của Hướng dẫn này).
– Lấy đúng lọ vắc xin và dung môi liên quan tới tai biến sau tiêm chủng, nếu đã dùng hết thì lấy vỏ lọ. Trong trường hợp không xác định được vỏ lọ dùng tiêm chủng cho đối tượng thì không lấy.
– Lấy thêm vắc xin cùng loại, cùng số lô, hạn dùng, cùng địa điểm xảy ra tai biến nặng sau tiêm chủng với số lọ đủ để kiểm tra an toàn, tối thiểu là 15 ml hoặc theo hướng dẫn của Viện Kiểm định quốc gia vắc xin và sinh phẩm y tế.
– Đối với vắc xin đông khô cần lấy thêm dung môi.
– Mẫu vắc xin nghi ngờ gửi tới Viện Kiểm định quốc gia vắc xin và sinh phẩm y tế phải bảo quản trong dây chuyền lạnh (như đối với các vắc xin được cấp phát cho các đơn vị để sử dụng) trong suốt quá trình vận chuyển.
– Trên mẫu gửi ghi rõ ngày, tháng, địa điểm xảy ra tai biến nặng sau tiêm chủng.
– Gửi mẫu kèm theo phiếu điều tra tai biến nặng sau tiêm chủng.
4.3.4. Lấy mẫu bệnh phẩm
– Trong trường hợp cần tiến hành giám định pháp y, thực hiện theo các quy định về giám định pháp y.
– Thu thập mẫu bệnh phẩm để kiểm tra nhiễm trùng, miễn dịch, mô bệnh học và vi rút học theo hướng dẫn của các phòng xét nghiệm liên quan và theo hướng dẫn hiện hành (xem phụ lục 16 của Hướng dẫn này).
4.4. Phân tích kết quả điều tra tai biến nặng sau tiêm chủng
4.4.1. Nhập số liệu theo các biến
Địa điểm, con người, thời gian, loại vắc xin và các triệu chứng theo hướng dẫn hiện hành (xem phụ lục 17 của Hướng dẫn này).
4.4.2. Thống kê số liệu
– Thống kê số trường hợp tai biến nặng sau tiêm chủng theo loại vắc xin.
– Thống kê nguyên nhân tai biến (dựa trên kết luận của Hội đồng cấp tỉnh và Hội đồng cấp Bộ).
– Thống kê số trường hợp đã tổ chức họp Hội đồng, thời gian điều tra, thời gian họp Hội đồng kể từ khi phát hiện tai biến sau nặng sau tiêm chủng.
– Thống kê các số liệu khác khi cần thiết.
4.4.3. So sánh, đánh giá kết quả
So sánh kết quả về tỷ lệ phản ứng thông thường, tai biến nặng sau tiêm chủng của từng loại vắc xin với tỷ lệ ước tính của các phân tích trước đó hoặc theo thống kê của Tổ chức Y tế Thế giới (xem phụ lục 18 của Hướng dẫn này).
4.5. Đánh giá nguyên nhân tai biến nặng sau tiêm chủng
Đánh giá nguyên nhân là sự xem xét một cách có hệ thống các thông tin về trường hợp tai biến nặng sau tiêm chủng để xác định mối liên quan giữa các phản ứng và tiêm chủng để:
– Xác định các vấn đề liên quan tới vắc xin.
– Xác định các vấn đề liên quan tới lỗi của dịch vụ tiêm chủng. – Loại trừ các trường hợp trùng hợp ngẫu nhiên.
4.5.1. Các trường hợp cần đánh giá nguyên nhân
– Trường hợp tai biến nặng sau tiêm chủng.
– Chùm phản ứng vượt quá số lượng thống kê về phản ứng chung của Tổ chức Y tế Thế giới hoặc có mức độ nghiêm trọng ảnh hưởng tới cộng đồng.
4.5.2. Đánh giá nguyên nhân và phân loại các trường hợp tai biến nặng sau tiêm chủng
– Đánh giá nguyên nhân tai biến nặng sau tiêm chủng theo hướng dẫn hiện hành (xem phụ lục 19 của Hướng dẫn này).
– Phân loại các trường hợp dựa vào những thông tin ghi nhận:
+ Các trường hợp có đầy đủ thông tin để kết luận nguyên nhân có thể được phân loại như sau:
A. Có liên quan tới tiêm chủng
A1: Liên quan tới đặc tính cố hữu của vắc xin
A2: Liên quan tới việc vắc xin không đạt chất lượng A3: Liên quan tới thực hành tiêm chủng
A4: Liên quan tới những lo sợ do bị tiêm chủng.
B. Chưa xác định
B1: Có mối liên quan tạm thời tới tiêm chủng nhưng chưa có đủ bằng chứng để kết luận (có thể do vắc xin mới), phải tiến hành điều tra thêm.
B2: Không xác định được nguyên nhân.
C. Không liên quan tới tiêm chủng do trùng hợp ngẫu nhiên hoặc do nguyên nhân khác.
+ Các trường hợp không có đủ thông tin để kết luận nguyên nhân được coi là “không phân loại được” và cần phải thu thập thêm thông tin để đánh giá nguyên nhân.
– Sử dụng sơ đồ phân loại nguyên nhân theo hướng dẫn hiện hành (xem phụ lục 20 của Hướng dẫn này).
CHƯƠNG 5.
HOẠT ĐỘNG CẢNH GIÁC DƯỢC
TRONG CÁC CHƯƠNG TRÌNH Y TẾ QUỐC GIA
5.1. Cảnh giác dược trong các Chương trình y tế Quốc gia
5.1.1. Sự cần thiết triển khai hoạt động Cảnh giác dược
Các chương trình y tế quốc gia tại Việt Nam được triển khai trong bối cảnh tỷ lệ mắc các bệnh HIV/AIDS, lao, sốt rét và các bệnh truyền nhiễm khác còn ở mức khá cao và chưa được khống chế đầy đủ hoặc thanh toán hoàn toàn. Các thuốc được sử dụng trong các chương trình y tế quốc gia có những đặc điểm riêng biệt về tác dụng phụ, độc tính nên cần được kiểm soát chặt chẽ để đảm bảo chất lượng và hiệu quả của chương trình. Khi sử dụng thuốc trên số lượng lớn người bệnh với các đối tượng khác nhau như trẻ em, người cao tuổi, phụ nữ mang thai, người suy giảm miễn dịch, việc sử dụng thuốc cần được điều chỉnh cho phù hợp và đảm bảo an toàn. Mặt khác, do yêu cầu cấp thiết trong kiểm soát dịch bệnh và tình trạng tăng kháng thuốc của vi sinh vật, các chương trình y tế quốc gia có thể phải sử dụng nhiều thuốc mới chưa được đánh giá đầy đủ và toàn diện về tính an toàn, đặc biệt trên quần thể người bệnh Việt Nam. Những tác động bất lợi đến hiệu quả của chương trình cũng có thể xảy ra nếu không giám sát một cách toàn diện để phát hiện, xử trí, đánh giá sớm các vấn đề liên quan đến an toàn thuốc sử dụng trong các chương trình y tế.
Vì vậy, việc triển khai các hoạt động Cảnh giác dược trong các chương trình y tế quốc gia là rất cần thiết để có những biện pháp dự phòng các phản ứng có hại có thể xảy ra đối với người bệnh, đặc biệt là các phản ứng có hại nghiêm trọng, ngoài dự kiến, góp phần đánh giá nguy cơ/lợi ích của thuốc và giúp cơ quan quản lý đưa ra các quyết định phù hợp.
5.1.2. Mối liên quan giữa các chương trình y tế quốc gia với hệ thống Cảnh giác dược
Cảnh giác dược và các chương trình y tế quốc gia có mối quan hệ mật thiết và hỗ trợ lẫn nhau. Các chương trình y tế quốc gia tham gia vào công tác phòng bệnh và chữa bệnh thông qua việc sử dụng thuốc cho cộng đồng, do đó có thể cung cấp một số lượng lớn người bệnh để tiến hành các hoạt động nghiên cứu, giám sát các vấn đề an toàn thuốc của Cảnh giác dược. Hơn nữa, các chương trình y tế quốc gia thường nhận được sự hỗ trợ của các tổ chức quốc tế và triển khai tốt các chương trình đào tạo cho nhân viên y tế. Đây là một thuận lợi giúp triển khai có hiệu quả hoạt động Cảnh giác dược trong các chương trình y tế quốc gia.
Ngược lại, Cảnh giác dược có thể hỗ trợ các chương trình y tế quốc gia thông qua việc cung cấp dữ liệu về độc tính và tính an toàn của các thuốc dùng trong chương trình, tạo cơ sở để khuyến cáo thay đổi hướng dẫn chẩn đoán và điều trị. Cảnh giác dược giúp xác định các yếu tố nguy cơ liên quan tới phản ứng có hại của thuốc, từ đó tăng cường việc sử dụng thuốc an toàn, hiệu quả trong các chương trình y tế quốc gia. Bên cạnh đó, lồng ghép Cảnh giác dược trong các chương trình y tế quốc gia còn giúp nhân viên y tế hình thành thói quen sử dụng thuốc hợp lý, an toàn.
Hệ thống Cảnh giác dược giám sát các thuốc sử dụng trong các chương trình y tế quốc gia như Chương trình phòng chống HIV/ AIDS, Chương trình chống lao quốc gia và Chương trình phòng chống sốt rét quốc gia đã được thiết lập tại nhiều nước trên thế giới và Việt Nam (hình 4).
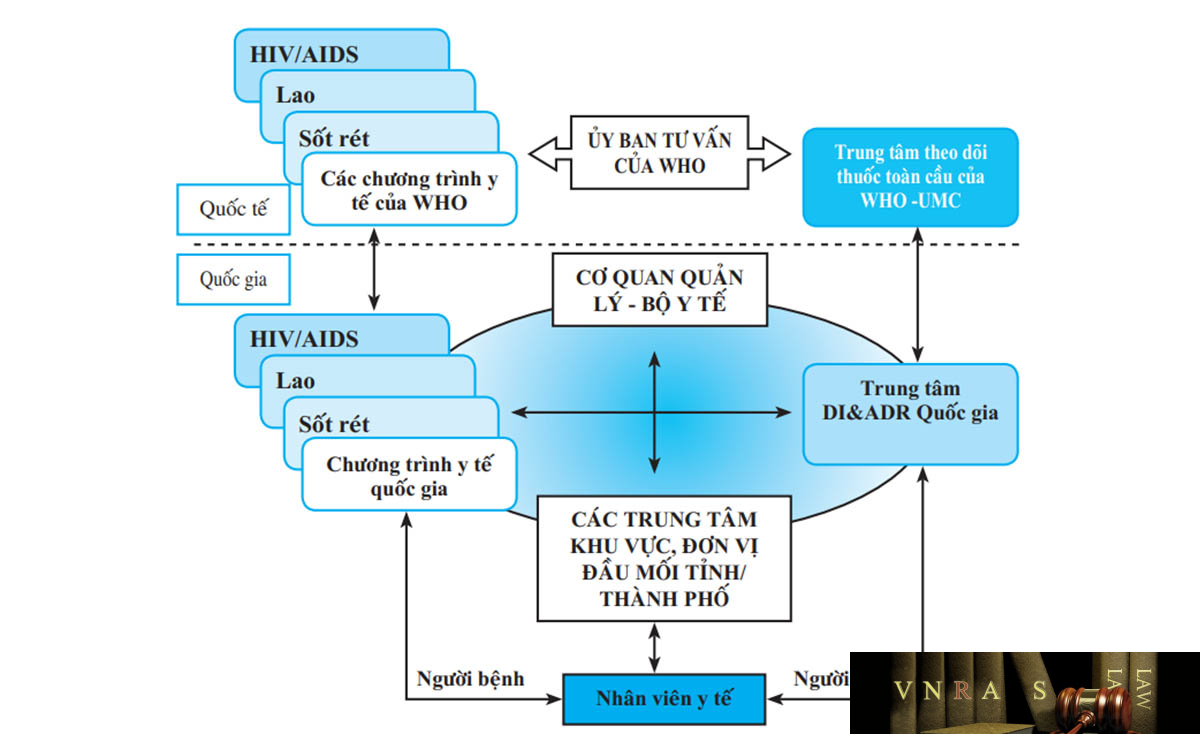
5.1.3. Mục tiêu của Cảnh giác dược trong các chương trình y tế quốc gia
– Xác định và giảm thiểu tỷ lệ phản ứng có hại của thuốc sử dụng trong chương trình.
– Đánh giá tác động của phản ứng có hại tới hiệu quả điều trị bao gồm bỏ trị, biến cố bất lợi để lại di chứng cho người bệnh, buộc người bệnh phải nhập viện để điều trị hoặc kéo dài thời gian nằm viện của người bệnh, đe dọa tính mạng, tử vong và gây dị tật bẩm sinh ở thai nhi.
– Xác định ảnh hưởng của các yếu tố như bệnh mắc kèm, các thuốc dùng đồng thời và thuốc có nguồn gốc dược liệu đến tỷ lệ xuất hiện, tính chất và mức độ nghiêm trọng của phản ứng có hại.
– Phát hiện các phản ứng có hại hiếm gặp hoặc phản ứng có hại xuất hiện khi sử dụng thuốc kéo dài, chưa được biết đến của thuốc.
5.1.4. Các phương pháp thu thập thông tin về tính an toàn của thuốc trong các chương trình y tế quốc gia
Các phương pháp thu thập thông tin về tính an toàn của thuốc có thể triển khai trong các chương trình y tế quốc gia bao gồm:
– Báo cáo tự nguyện
– Báo cáo tự nguyện có chủ đích
– Giám sát chủ động
(Xem thông tin chi tiết trong Chương 1 của tài liệu này).
Báo cáo tự nguyện và báo cáo tự nguyện có chủ đích có thể được áp dụng để theo dõi thường quy các biến cố bất lợi tại các cơ sở khám bệnh, chữa bệnh trong các chương trình y tế quốc gia. Trong đó, phương pháp báo cáo tự nguyện hướng đến thu thập tất cả các phản ứng có hại của tất cả các loại thuốc, còn phương pháp báo cáo tự nguyện có chủ đích chỉ tập trung theo dõi và báo cáo theo một số tiêu chí nhất định (như trên một nhóm người bệnh cụ thể, một số phản ứng có hại cụ thể của một số thuốc nhất định). Vì vậy, phương pháp báo cáo tự nguyện có chủ đích khá phù hợp để triển khai trong các chương trình y tế quốc gia do có thể nâng cao chất lượng báo cáo và giảm bớt khối lượng công việc cho nhân viên y tế hơn so với báo cáo tự nguyện.
Phương pháp giám sát chủ động có thiết kế và cách tiến hành tương tự một nghiên cứu thuần tập trong dịch tễ học, được sử dụng để phát hiện những biến cố bất lợi mới, đặc biệt thuốc mới hoặc để thu thập thông tin một cách toàn diện về các biến cố bất lợi đã biết. Phương pháp này cũng được áp dụng rộng rãi để ghi nhận một cách đầy đủ các biến cố bất lợi trong các chương trình y tế quốc gia.
5.2. Theo dõi phản ứng có hại của thuốc trong các chương trình y tế quốc gia
5.2.1. Chương trình chống Lao Quốc gia
a) Đối tượng thực hiện và phạm vi áp dụng
Phạm vi áp dụng: các cơ sở khám bệnh, chữa bệnh công lập và ngoài công lập có đăng ký tham gia điều trị lao; các cơ sở quản lý điều trị lao tại cộng đồng (tổ chống lao quận/huyện hoặc trạm y tế phường/xã đủ điều kiện chăm sóc và điều trị người bệnh lao).
Đối tượng tham gia báo cáo: bác sĩ, dược sĩ, điều dưỡng, giám sát viên tại cộng đồng (nhân viên y tế tổ chống lao quận/huyện hoặc trạm y tế phường/xã), người bệnh và người nhà người bệnh. Khuyến khích nhiều người cùng tham gia viết hoàn thiện báo cáo.
b) Báo cáo phản ứng có hại của thuốc trong chương trình chống Lao Quốc gia
Trong chương trình chống lao, việc phát hiện, xử trí, báo cáo và dự phòng các ADR cũng tuân theo những nguyên tắc giống như đối với tất cả các thuốc khác, đồng thời có những đặc thù riêng. Một số điểm sau cần lưu ý khi triển khai công tác báo cáo ADR trong chương trình chống lao:
Các trường hợp cần báo cáo
Sử dụng một bản báo cáo riêng cho mỗi một người bệnh mỗi khi phát hiện hay nghi ngờ xảy ra phản ứng có hại của thuốc. Tất cả các biến cố bất lợi xảy ra trong quá trình điều trị lao nghi ngờ là phản ứng có hại gây ra bởi các thuốc kháng lao và các thuốc sử dụng đồng thời đều cần được báo cáo. Các trường hợp ưu tiên báo cáo:
– Các phản ứng có hại nghiêm trọng (các phản ứng dẫn đến một trong những hậu quả sau: gây tử vong; đe dọa tính mạng; buộc người bệnh phải nhập viện để điều trị hoặc kéo dài thời gian nằm viện của người bệnh, cần phải thay đổi phác đồ điều trị; để lại di chứng nặng nề hoặc vĩnh viễn cho người bệnh làm cho người bệnh tự ý bỏ thuốc trong quá trình điều trị ngoại trú; hoặc bất kỳ phản ứng có hại được nhân viên y tế nhận định là gây ra hậu quả nghiêm trọng về mặt lâm sàng).
– Tất cả phản ứng có hại của các thuốc kháng lao mới đưa vào sử dụng trong điều trị.
– Phản ứng có hại mới chưa từng được biết đến của thuốc chống lao (chưa được mô tả trong tờ hướng dẫn sử dụng thuốc, Dược thư Quốc gia Việt Nam hay các tài liệu tham khảo thông tin thuốc khác).
– Phản ứng có hại xảy ra liên tục với một thuốc hoặc một lô thuốc trong một thời gian ngắn tại cơ sở khám bệnh, chữa bệnh có đăng ký điều trị lao.
Khuyến khích nhân viên y tế báo cáo các vấn đề về chất lượng thuốc và sai sót liên quan đến thuốc.
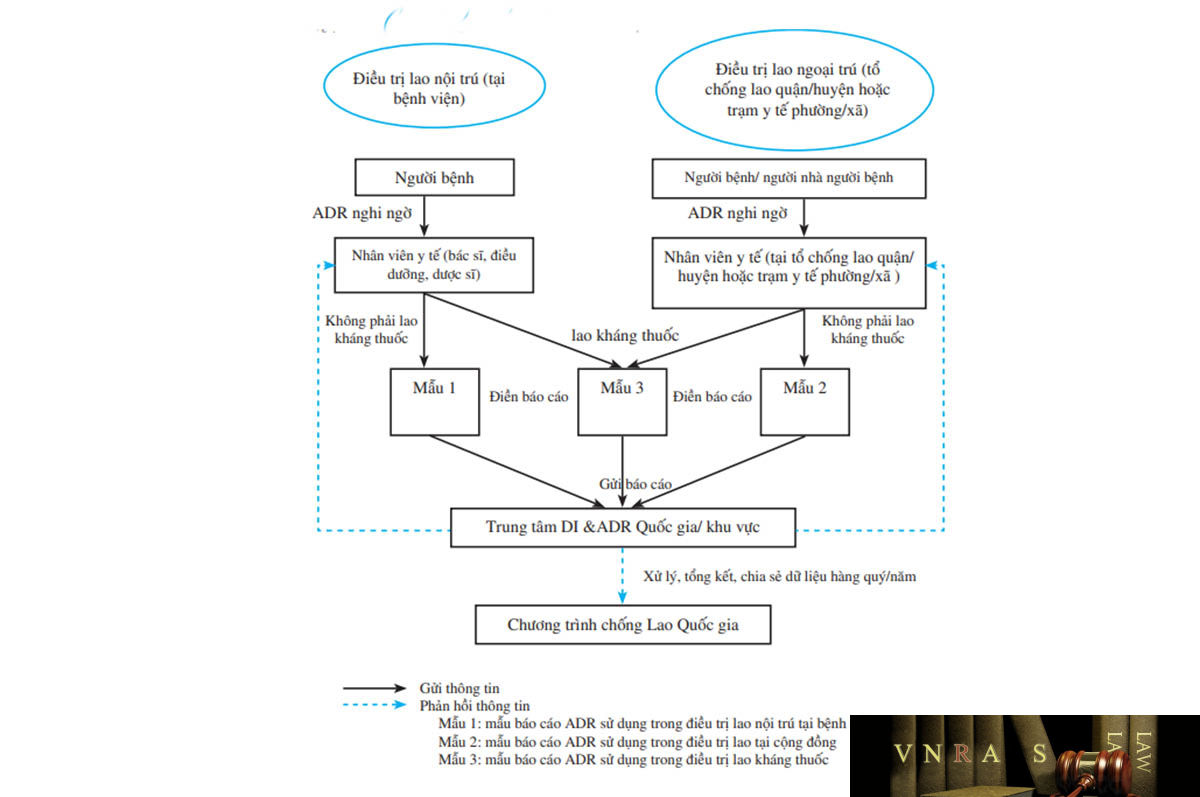
Thời gian gửi báo cáo
Báo cáo cần được gửi trong thời gian sớm nhất có thể sau khi xảy ra phản ứng, ngay cả khi thông tin thu được chưa đầy đủ (báo cáo ban đầu). Trong trường hợp này, có thể bổ sung báo cáo nếu thu thập được thêm thông tin (báo cáo bổ sung). Bảo đảm việc gửi báo cáo tới Trung tâm Quốc gia về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc đúng thời hạn:
– Báo cáo phản ứng có hại nghiêm trọng gây tử vong hoặc đe dọa tính mạng người bệnh: gửi trong thời gian sớm nhất có thể nhưng không muộn hơn 7 ngày làm việc kể từ thời điểm phát hiện ra phản ứng.
– Báo cáo phản ứng có hại nghiêm trọng khác: gửi trong thời gian sớm nhất có thể nhưng không muộn hơn 15 ngày làm việc kể từ thời điểm phát hiện ra phản ứng.
– Các phản ứng có hại khác có thể tập hợp gửi hàng tháng, trước ngày mùng 5 của tháng kế tiếp.
Quy trình báo cáo
Việc điền và gửi báo cáo nghi ngờ phản ứng có hại của thuốc kháng lao được thực hiện theo quy trình trong hình 5.
– Trong trường hợp xảy ra ADR ở người bệnh sử dụng phác đồ điều trị lao kháng thuốc kể cả điều trị nội trú và ngoại trú, nhân viên y tế có thể sử dụng mẫu báo cáo phản ứng có hại của thuốc chống lao sử dụng trong điều trị lao kháng thuốc (mẫu 3) để điền báo cáo theo hướng dẫn hiện hành (xem phụ lục 23 của Hướng dẫn này).
– Với những trường hợp xuất hiện ADR trên người bệnh điều trị lao nội trú không dùng phác đồ lao kháng thuốc, nhân viên y tế có thể báo cáo theo mẫu báo cáo phản ứng có hại của thuốc chống lao sử dụng trong điều trị lao nội trú tại bệnh viện (mẫu 1) theo hướng dẫn hiện hành (xem phụ lục 21 của Hướng dẫn này).
– Trường hợp người bệnh đang điều trị lao ngoại trú không dùng phác đồ lao kháng thuốc, thông tin về ADR thu thập được có thể được nhân viên y tế của tại tổ chống lao quận/huyện hoặc trạm y tế phường/xã báo cáo theo mẫu báo cáo phản ứng có hại của thuốc chống lao sử dụng trong điều trị lao tại cộng đồng (mẫu 2) theo hướng dẫn hiện hành (xem phụ lục 22 của Hướng dẫn này).
Tất cả các báo cáo cần được hoàn thành trong thời gian sớm nhất có thể và gửi về Trung tâm Quốc gia về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc. Trung tâm sẽ xử lý, đánh giá từng báo cáo và gửi phản hồi tới người báo cáo trong một số trường hợp đặc biệt. Dữ liệu về những báo cáo ADR liên quan đến thuốc kháng lao sẽ được tổng hợp và thông báo định kỳ cho Chương trình chống lao Quốc gia.
5.2.2. Chương trình phòng, chống HIV/AIDS
a) Đối tượng thực hiện và phạm vi áp dụng
Phạm vi áp dụng: các cơ sở y tế, cơ sở khám bệnh, chữa bệnh có triển khai điều trị bằng thuốc ARV (sau đây gọi tắt là cơ sở điều trị); các Trung tâm phòng, chống HIV/AIDS các tỉnh/thành phố và Ủy ban Phòng, chống AIDS thành phố Hồ Chí Minh.
Đối tượng thực hiện: bác sĩ, y sĩ, dược sĩ, dược tá, điều dưỡng, nhân viên tư vấn và các nhân viên y tế khác trong chương trình phòng, chống HIV/AIDS (gọi chung là nhân viên y tế).
b) Báo cáo phản ứng có hại của thuốc trong chương trình phòng, chống HIV/AIDS
Nội dung báo cáo
– Báo cáo đơn lẻ: báo cáo tất cả các biến cố bất lợi xảy ra trong quá trình điều trị nghi ngờ là phản ứng có hại gây ra bởi thuốc ARV hoặc các thuốc dùng đồng thời cho người bệnh nhiễm HIV/AIDS theo quy định hiện hành (xem phụ lục 24 của Hướng dẫn này). Ưu tiên báo cáo các trường hợp sau:
+ Phản ứng có hại nghiêm trọng ở mức độ 3 và mức độ 4 theo phân loại mức độ nặng của phản ứng có hại của thuốc trong Hướng dẫn chẩn đoán và điều trị HIV/AIDS hiện hành của Bộ Y tế.
+ Bất kỳ phản ứng nào dẫn đến một trong những hậu quả: thay đổi phác đồ điều trị, bỏ trị, ngừng điều trị hoặc cần phải can thiệp y khoa để xử trí phản ứng có hại.
+ Bất kỳ phản ứng có hại được nhân viên y tế nhận định là gây ra hậu quả nghiêm trọng về mặt lâm sàng.
+ Tất cả phản ứng có hại của các thuốc mới hoặc phác đồ mới.
+ Phản ứng có hại mới chưa được ghi nhận với thuốc (chưa được mô tả trong Hướng dẫn chẩn đoán và điều trị HIV/AIDS của Bộ Y tế, tờ hướng dẫn sử dụng thuốc, Dược thư Quốc gia Việt Nam hay các tài liệu tham khảo thông tin thuốc khác). Các trường hợp phản ứng có hại nghiêm trọng, nhiều khả năng do sai sót, phản ứng gây quan tâm cho cha mẹ và cộng đồng, phản ứng nhẹ nhưng có tần số xuất hiện bất thường, cụm phản ứng sau tiêm chủng phải báo cáo trong vòng 24 giờ kể từ khi ghi nhận thông tin và báo cáo hàng tuần về diễn biến điều tra, xử lý.
– Báo cáo định kỳ: báo cáo hàng tháng về các phản ứng có hại của thuốc ARV theo quy định hiện hành (xem phụ lục 25 và phụ lục 26 của Hướng dẫn này).
Tổ chức thực hiện
– Cục Phòng, chống HIV/AIDS
+ Thực hiện tiếp nhận báo cáo định kỳ từ các tỉnh/thành phố và báo cáo (tổng hợp, phân tích, khuyến nghị) từ Trung tâm Quốc gia về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc.
+ Phối hợp với các đơn vị đưa ra khuyến nghị, xử trí (nếu có) đối với các thuốc ARV xuất hiện các phản ứng có hại tùy mức độ xử trí.
+ Thực hiện kiểm tra giám sát việc thực hiện triển khai hoạt động theo dõi phản ứng có hại của thuốc tại các tỉnh/thành phố.
– Trung tâm Quốc gia về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc
+ Tiếp nhận, phân tích, phản hồi báo cáo đơn lẻ và báo cáo định kỳ từ các cơ sở điều trị. Thực hiện phản hồi nhanh thông tin tới các đơn vị liên quan đối với các phản ứng có hại nghiêm trọng.
+ Định kỳ hàng tháng thực hiện tổng hợp, phân tích báo cáo phản ứng có hại của thuốc ARV, đưa ra khuyến nghị và phản hồi về Cục Phòng, chống HIV/AIDS và các đơn vị có liên quan.
– Trung tâm Phòng, chống HIV/AIDS các tỉnh/thành phố và Ủy ban Phòng, chống HIV/AIDS thành phố Hồ Chí Minh
+ Định kỳ hàng tháng, tổng hợp báo cáo định kỳ của các cơ sở điều trị trên địa bàn tỉnh/thành phố gửi Cục phòng, chống HIV/ AIDS trước ngày 10 của tháng kế tiếp theo quy định hiện hành (xem phụ lục 26 của Hướng dẫn này).
+ Thực hiện giám sát, hỗ trợ thực hiện báo cáo đơn lẻ và báo cáo định kỳ phản ứng có hại của thuốc tại các cơ sở điều trị trên địa bàn theo qui định.
– Cơ sở điều trị
+ Thực hiện báo cáo đơn lẻ phản ứng có hại của thuốc theo quy định hiện hành (xem phụ lục 24 của Hướng dẫn này) cho Trung tâm Quốc gia về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc theo qui định thời gian như sau:
● Phản ứng có hại nghiêm trọng gây tử vong hoặc đe dọa tính mạng người bệnh (mức độ 4): gửi báo cáo trong thời gian sớm nhất có thể nhưng không muộn hơn 7 ngày làm việc kể từ thời điểm phát hiện ra phản ứng.
● Phản có hại nghiêm trọng mức độ 3: gửi báo cáo trong thời gian sớm nhất có thể nhưng không muộn hơn 15 ngày làm việc kể từ thời điểm phát hiện ra phản ứng.
● Các phản ứng có hại khác có thể tập hợp gửi hàng tháng, trước ngày mùng 5 của tháng kế tiếp.
● Báo cáo cần được gửi trong thời gian sớm nhất có thể sau khi xảy ra phản ứng, ngay cả khi thông tin thu được chưa đầy đủ (báo cáo ban đầu). Trong trường hợp này, có thể bổ sung báo cáo nếu thu thập được thêm thông tin (báo cáo bổ sung).
+ Định kỳ hàng tháng thực hiện báo cáo định kỳ tại cơ sở điều trị và báo cáo Trung tâm phòng, chống HIV/AIDS tỉnh/thành phố trước ngày 5 của tháng kế tiếp theo quy định hiện hành (xem phụ lục 25 của Hướng dẫn này) .
5.2.3. Chương trình phòng chống Sốt rét Quốc gia
a) Đối tượng thực hiện và phạm vi áp dụng
Đối tượng thực hiện: tất cả các nhân viên y tế bao gồm: bác sĩ, y sĩ, dược sĩ, dược tá, điều dưỡng viên, hộ sinh viên, kỹ thuật viên và các nhân viên y tế khác tại các cơ sở khám bệnh, chữa bệnh có sử dụng thuốc điều trị và dự phòng sốt rét.
Phạm vi áp dụng: các cơ sở khám bệnh, chữa bệnh có điều trị người bệnh sốt rét, các Trung tâm Phòng chống Sốt rét, Ký sinh trùng, Côn trùng, Trung tâm Y tế dự phòng tỉnh/thành phố trên cả nước.
b) Quy trình báo cáo phản ứng có hại của thuốc trong chương trình phòng chống Sốt rét Quốc gia
Các trường hợp cần báo cáo
Báo cáo tất cả các biến cố bất lợi xảy ra trong quá trình điều trị nghi ngờ là phản ứng có hại do thuốc điều trị sốt rét gây ra hoặc các thuốc dùng đồng thời cho người bệnh bị sốt rét. Ưu tiên báo cáo các trường hợp sau:
– Phản ứng có hại nghiêm trọng: bất cứ phản ứng có hại nào dẫn đến một trong các hậu quả sau: gây tử vong; đe dọa tính mạng; buộc người bệnh phải nhập viện để điều trị hoặc kéo dài thời gian nằm viện của người bệnh; cần phải thay đổi phác đồ điều trị; để lại di chứng nặng nề hoặc vĩnh viễn cho người bệnh; gây dị tật bẩm sinh; hoặc bất kỳ phản ứng có hại được nhân viên y tế nhận định là gây ra hậu quả nghiêm trọng về mặt lâm sàng.
– Phản ứng có hại mới chưa từng được biết đến của thuốc sốt rét (chưa được mô tả trong Hướng dẫn chẩn đoán, điều trị và dự phòng bệnh sốt rét của Bộ Y tế, tờ hướng dẫn sử dụng thuốc, Dược thư Quốc gia Việt Nam hay các tài liệu tham khảo thông tin thuốc khác).
– Phản ứng có hại xảy ra liên tục với một thuốc hoặc một lô thuốc trong một thời gian ngắn tại cơ sở khám bệnh, chữa bệnh.
Khuyến khích nhân viên y tế báo cáo các vấn đề về chất lượng thuốc và sai sót liên quan đến thuốc.
Nội dung báo cáo
Điền đầy đủ thông tin vào mẫu báo cáo ADR của thuốc dùng trong điều trị sốt rét theo hướng dẫn hiện hành (xem phụ lục 27 của Hướng dẫn này), bao gồm các thông tin:
– Tên cơ sở điều trị, mã báo cáo của đơn vị (nếu có);
– Thông tin người bệnh: họ tên, ngày sinh hoặc tuổi, giới tính, cân nặng, nhóm dân tộc, tiền sử dị ứng, nghiện thuốc lá, nghiện rượu, có mắc kèm theo các bệnh gan, thận hay không, kết quả xét nghiệm ký sinh trùng sốt rét;
– Thông tin về phản ứng có hại: ngày xuất hiện phản ứng, thời điểm xuất hiện phản ứng, các biểu hiện và diễn biến của phản ứng, các xét nghiệm liên quan phản ứng, mức độ nghiêm trọng của phản ứng, cách xử trí và kết quả sau khi xử trí phản ứng;
– Thuốc nghi ngờ gây phản ứng có hại: trong số các thuốc điều trị sốt rét đã được liệt kê sẵn tên thuốc, dạng dùng và hàm lượng, điền bổ sung các thông tin liên quan đến thuốc sốt rét nghi ngờ gây ADR (tên nhà sản xuất thuốc, số lô, hạn dùng, liều dùng, đường dùng thuốc, thời điểm bắt đầu và kết thúc dùng thuốc); sau khi ngừng/giảm liều phản ứng có cải thiện không, tái sử dụng thuốc nghi ngờ phản ứng có xuất hiện lại không; thông tin thuốc dùng đồng thời (tên thuốc, dạng bào chế, hàm lượng, đường dùng, liều dùng một lần, số lần dùng 1 ngày, tổng liều, lý do dùng thuốc, thời điểm bắt đầu và kết thúc dùng thuốc);
– Thông tin về người báo cáo (họ tên, nghề nghiệp/chức vụ, điện thoại liên lạc, email, chữ ký, dạng báo cáo và ngày báo cáo).
Quy trình báo cáo
Các cơ sở điều trị, phòng khám ngoại trú, Trung tâm Phòng chống Sốt rét, Ký sinh trùng và Côn trùng/Trung tâm Y tế dự phòng tỉnh/thành phố báo cáo trực tiếp cho Trung tâm Quốc gia về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc theo qui định thời gian như sau:
– Báo cáo cần được gửi trong thời gian sớm nhất có thể sau khi xảy ra phản ứng, ngay cả khi thông tin thu được chưa đầy đủ (báo cáo ban đầu). Trong trường hợp này, có thể bổ sung báo cáo nếu thu thập được thêm thông tin (báo cáo bổ sung).
– Bảo đảm việc gửi báo cáo tới Trung tâm Quốc gia về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc đúng thời hạn:
+ Báo cáo phản ứng có hại nghiêm trọng gây tử vong hoặc đe dọa tính mạng người bệnh: gửi trong thời gian sớm nhất có thể nhưng không muộn hơn 7 ngày làm việc kể từ thời điểm phát hiện ra phản ứng.
+ Báo cáo phản ứng có hại nghiêm trọng khác: gửi trong thời gian sớm nhất có thể nhưng không muộn hơn 15 ngày làm việc kể từ thời điểm phát hiện ra phản ứng.
+ Các phản ứng có hại khác có thể tập hợp gửi hàng tháng, trước ngày mùng 5 của tháng kế tiếp.
CHƯƠNG 6.
HOẠT ĐỘNG CẢNH GIÁC DƯỢC
TRONG HỆ THỐNG CUNG ỨNG THUỐC
Thực hành Cảnh giác dược trong hệ thống cung ứng thuốc là nhiệm vụ quan trọng, không thể tách rời khỏi hệ thống Cảnh giác dược. Chương này mô tả ngắn gọn trách nhiệm liên quan đến hoạt động Cảnh giác dược của các đơn vị thuộc hệ thống cung ứng thuốc bao gồm:
– Các đơn vị kinh doanh thuốc;
– Các cơ sở bán lẻ thuốc.
A. Hoạt động Cảnh giác dược tại đơn vị kinh doanh thuốc
6.1. Nhân viên phụ trách hoạt động Cảnh giác dược
– Để hoạt động Cảnh giác dược được thực hiện tốt, đơn vị kinh doanh thuốc cần bổ nhiệm nhân viên phụ trách hoạt động Cảnh giác dược có kiến thức về lý thuyết và kỹ năng thực hành trong lĩnh vực Cảnh giác dược. Nhân viên này cần được đào tạo về chuyên môn, kỹ năng quản lý hệ thống để có thể triển khai các hoạt động Cảnh giác dược trước khi chính thức đảm nhiệm vị trí. Trong trường hợp cần thiết, đơn vị kinh doanh thuốc có thể xem xét đào tạo bổ sung kiến thức về các thuốc nằm trong hệ thống Cảnh giác dược của doanh nghiệp cho nhân viên phụ trách hoạt động Cảnh giác dược.
– Nếu nhân viên phụ trách hoạt động Cảnh giác dược đang trong quá trình đào tạo về các kiến thức và kỹ năng nói trên, đơn vị kinh doanh thuốc cần phân công một nhân viên khác đã được đào tạo trong lĩnh vực này hỗ trợ cho nhân viên đó. Việc bổ nhiệm nhân viên phụ trách hoạt động Cảnh giác dược cũng như nhân viên hỗ trợ (nếu có) nên được chính thức hóa bằng văn bản.
– Đơn vị kinh doanh thuốc cần đánh giá năng lực của nhân viên phụ trách hoạt động Cảnh giác dược trước khi giao nhiệm vụ bao gồm: kiểm tra chứng chỉ chuyên môn, kiến thức liên quan đến các yêu cầu về Cảnh giác dược ở Việt Nam cũng như kinh nghiệm trong hoạt động Cảnh giác dược, …
– Nhiệm vụ của nhân viên phụ trách hoạt động Cảnh giác dược:
+ Là đầu mối liên lạc với Cục Quản lý Dược và các cơ quan quản lý nhà nước liên quan trong lĩnh vực y tế, Trung tâm Quốc gia và Trung tâm khu vực về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc để trao đổi các vấn đề Cảnh giác dược của đơn vị.
+ Tập hợp thông tin về ADR và các biến cố bất lợi nghi ngờ liên quan đến thuốc của đơn vị mình từ các nhân viên y tế, cơ sở khám bệnh, chữa bệnh, cộng đồng, các nhân viên khác trong đơn vị để gửi về Trung tâm Quốc gia hoặc Trung tâm khu vực về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc theo theo quy định hiện hành.
+ Cập nhật thông tin về tính an toàn của thuốc theo quyết định của các cơ quan quản lý dược trên thế giới.
+ Tập huấn và hướng dẫn cho nhân viên của đơn vị về các quy định quản lý nhà nước trong lĩnh vực Cảnh giác dược, các quy trình hoạt động Cảnh giác dược của đơn vị, thông tin cập nhật về tính an toàn liên quan đến các sản phẩm thuốc của đơn vị đang lưu hành tại Việt Nam.
+ Các nhiệm vụ khác theo phân công của đơn vị.
6.2. Trách nhiệm của đơn vị kinh doanh thuốc trong thực hành Cảnh giác dược
6.2.1. Báo cáo phản ứng có hại của thuốc
a) Báo cáo ADR đơn lẻ xảy ra trên lãnh thổ Việt Nam
– Các trường hợp phải báo cáo: tất cả phản ứng có hại của thuốc xảy ra trên lãnh thổ Việt Nam liên quan đến thuốc do đơn vị sản xuất, đăng ký hoặc phân phối.
– Với các biến cố bất lợi nghiêm trọng xảy ra trong thử nghiệm lâm sàng trên lãnh thổ Việt Nam, thực hiện theo hướng dẫn tại chương 7 của tài liệu này.
– Yêu cầu:
+ Báo cáo cần được gửi trong thời gian sớm nhất có thể sau ngày số không, ngay cả khi thông tin thu được chưa đầy đủ (báo cáo ban đầu). Trong trường hợp này, cần gửi bổ sung báo cáo nếu thu thập được thêm thông tin (báo cáo bổ sung).
+ Báo cáo ban đầu: bao gồm tối đa các thông tin hiện có, trong đó, cần có các thông tin tối thiểu đủ để xác định rõ người bệnh, người báo cáo, phản ứng xảy ra và thuốc nghi ngờ.
+ Báo cáo bổ sung: cập nhật, chỉnh sửa các thông tin chưa có, chưa đầy đủ hoặc chưa chính xác trong báo cáo ban đầu liên quan đến người bệnh, phản ứng xảy ra, thuốc nghi ngờ, người báo cáo, thuốc dùng đồng thời, cách xử trí phản ứng, đánh giá của bác sĩ điều trị hoặc người báo cáo, …
– Thời hạn báo cáo:
+ ADR nghiêm trọng gây tử vong hoặc đe dọa tính mạng người bệnh: báo cáo ban đầu gửi trong thời gian sớm nhất có thể nhưng không muộn hơn 7 ngày làm việc kể từ ngày số không; báo cáo bổ sung gửi trong thời gian sớm nhất có thể nhưng không muộn hơn 15 ngày làm việc kể từ ngày nhận được thông tin bổ sung.
+ ADR nghiêm trọng không thuộc loại gây tử vong hoặc đe dọa tính mạng người bệnh: báo cáo ban đầu gửi trong thời gian sớm nhất có thể nhưng không muộn hơn 15 ngày làm việc kể từ ngày số không; báo cáo bổ sung gửi trong thời gian sớm nhất có thể nhưng không muộn hơn 15 ngày làm việc kể từ ngày nhận được thông tin bổ sung.
+ ADR không nghiêm trọng: tập hợp gửi hàng tháng, trước ngày 15 của tháng kế tiếp.
– Biểu mẫu báo cáo: có thể sử dụng một trong hai mẫu báo cáo sau:
+ Mẫu báo cáo phản ứng có hại của Bộ Y tế dành cho các cơ sở khám bệnh, chữa bệnh (xem phụ lục 1 của Hướng dẫn này).
+ Mẫu báo cáo của Hội đồng các tổ chức quốc tế về khoa học y học (mẫu báo cáo CIOMS) (xem phụ lục 28 của Hướng dẫn này)
– Hình thức báo cáo: gửi báo cáo qua đường bưu điện, fax hoặc email.
b) Báo cáo ADR định kỳ
– Các trường hợp cần báo cáo: tất cả các trường hợp nghi ngờ xảy ra ADR trong và ngoài lãnh thổ Việt Nam, liên quan đến mỗi thuốc mà đơn vị sản xuất, đăng ký hoặc phân phối ở Việt Nam.
– Các biểu mẫu cần báo cáo:
+ Có thể sử dụng một trong hai mẫu báo cáo sau: Báo cáo định kỳ về tính an toàn của thuốc (Periodic Safety Update Report, PSUR) hoặc Báo cáo đánh giá định kỳ về hiệu quả và tính an toàn của thuốc (Periodic Benefit Risk Evaluation Report, PBRER) theo Hướng dẫn E2C của Hội nghị hòa hợp quốc tế (International Conference on Harmonisation, ICH) tại website www.ich.org (báo cáo có thể được viết bằng ngôn ngữ tiếng Anh hoặc tiếng Việt). Báo cáo này cần nộp kèm báo cáo tóm tắt về hiệu quả và tính an toàn của thuốc bằng tiếng Việt (xem phụ lục 29 của Hướng dẫn này).
+ Trong trường hợp không có Báo cáo PSUR hoặc PBRER, đơn vị kinh doanh thuốc có thể nộp Báo cáo an toàn, hiệu quả của thuốc sau khi lưu hành theo quy định hiện hành về đăng ký thuốc để thay thế (xem phụ lục 30 và phụ lục 31 của Hướng dẫn này).
+ Bản sao thông tin Tóm tắt đặc tính sản phẩm, tờ Hướng dẫn sử dụng thuốc và/hoặc tờ Thông tin dành cho người bệnh cập nhật tại Việt Nam.
– Thời hạn báo cáo: trong vòng 15 ngày làm việc sau khi đơn vị tổng hợp xong thông tin theo chu kỳ của từng sản phẩm nhưng không muộn hơn 90 ngày theo lịch sau khoảng thời gian mà báo cáo bao phủ. Trong đó, chu kỳ sản phẩm do đơn vị kinh doanh thuốc lựa chọn và đăng ký khi nộp báo cáo ADR định kỳ lần đầu. Hướng dẫn này khuyến khích báo cáo theo chu kỳ hàng năm kể từ ngày sinh quốc tế của thuốc.
– Phương thức báo cáo: ưu tiên gửi báo cáo bằng bản điện tử qua đĩa CD hoặc email.
Lưu ý: đối với thuốc được cấp số đăng ký có yêu cầu theo dõi, đánh giá an toàn, hiệu quả trong quá trình lưu hành, đơn vị kinh doanh thuốc cần thực hiện chế độ báo cáo theo quy định hiện hành về đăng ký thuốc.
c) Nơi nhận báo cáo
– Trung tâm Quốc gia về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc (nhận báo cáo từ tất cả các tỉnh/thành phố trên phạm vi toàn quốc)
Địa chỉ: Trường Đại học Dược Hà Nội, 13-15 Lê Thánh Tông, Quận Hoàn Kiếm, Hà Nội
Điện thoại: (04) 3933 5618
Fax: (04) 3933 5642
E-mail: di.pvcenter@gmail.com
Trang thông tin điện tử: http://canhgiacduoc.org.vn
– Trung tâm khu vực về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc thành phố Hồ Chí Minh (nhận báo cáo của các tỉnh/thành phố từ Đà Nẵng trở vào)
Địa chỉ: Bệnh viện Chợ Rẫy, 201B Nguyễn Chí Thanh, Phường 12, Quận 5, Thành phố Hồ Chí Minh
Điện thoại: (08) 3855 4137- Ext: 794 hoặc (08) 3856 3537 Fax: (08) 3856 3537
E-mail: adrhcm@choray.vn
6.2.2. Cập nhật thông tin liên quan đến tính an toàn của thuốc
a) Trong trường hợp thuốc có số đăng ký lưu hành tại Việt Nam cũng được lưu hành ở nước ngoài, đơn vị kinh doanh thuốc cần cập nhật các thông tin mới liên quan đến tính an toàn của thuốc dẫn đến thay đổi về quản lý thuốc như sửa đổi thông tin Tóm tắt đặc tính sản phẩm, tạm ngừng sử dụng, thu hồi, rút số đăng ký, … ở bất kỳ quốc gia nào khác mà thuốc được phép lưu hành.
– Thời hạn cập nhật: trong vòng 3 ngày làm việc kể từ khi đơn vị ghi nhận được thông tin.
– Hình thức cập nhật: cập nhật thông tin bằng công văn chính thức, gửi kèm tài liệu gốc về thông tin cập nhật liên quan đến tính an toàn của thuốc.
– Nơi nhận báo cáo về thông tin cập nhật liên quan đến tính an toàn của thuốc:
Cục Quản lý Dược – Bộ Y tế
Địa chỉ: 138A Giảng Võ, Hà Nội
Điện thoại: (84-4) 3736 6483
Fax: (84-4) 3823 4758
Email: cqldvn@moh.gov.vn
b) Đơn vị kinh doanh thuốc có trách nhiệm cập nhật thông tin về chất lượng, an toàn và hiệu quả của thuốc do cơ sở mình sản xuất, đăng ký, kinh doanh trong trường hợp các thông tin này chưa được cập nhật vào hồ sơ đăng ký thuốc khi thuốc còn đang lưu hành trên thị trường. Việc cập nhật này được thực hiện theo quy định hiện hành về đăng ký thuốc.
6.2.3. Lập Kế hoạch quản lý nguy cơ và cập nhật các thay đổi về cân bằng nguy cơ/lợi ích
Tại thời điểm được cấp phép lưu hành, thông tin về tính an toàn của một thuốc còn tương đối hạn chế. Nguyên nhân là do hạn chế trong các thử nghiệm lâm sàng như số lượng đối tượng nghiên cứu tương đối nhỏ so với nhóm người bệnh đích, đối tượng nghiên cứu hạn chế về độ tuổi, giới tính, chủng tộc, bệnh mắc kèm, thuốc dùng đồng thời, điều kiện dùng thuốc; thời gian dùng thuốc và quá trình theo dõi người bệnh tương đối ngắn. Do đó, không phải tất cả những nguy cơ thực tế hoặc tiềm ẩn đều đã được xác định ở thời điểm xét duyệt cấp số đăng ký thuốc. Nhiều nguy cơ liên quan tới việc sử dụng thuốc chỉ được phát hiện và mô tả sau khi thuốc được phép lưu hành. Phần này sẽ trình bày những khái niệm cơ bản để xây dựng Kế hoạch quản lý nguy cơ (Risk Management Plan, RMP), bao gồm kế hoạch giảm thiểu nguy cơ (Risk Minimisation Plan), thông tin về tính an toàn (safety specification) và kế hoạch Cảnh giác dược (Pharmacovigilance Plan).
a) Các bước trong quy trình quản lý nguy cơ
– Phát hiện nguy cơ (risk identification);
– Phân tích nguy cơ (risk characterisation);
– Đánh giá cân bằng nguy cơ/lợi ích (risk/benefit assessment);
– Giảm thiểu nguy cơ và truyền thông nguy cơ (risk minimisation and communication);
– Đánh giá hiệu quả (effectiveness measurement).
b) Thực hành quản lý nguy cơ tại đơn vị kinh doanh thuốc
Xác định và phân loại thông tin về tính an toàn
Mục đích của thông tin về tính an toàn là cung cấp thông tin tóm tắt về hồ sơ an toàn của thuốc. Thông tin về tính an toàn nên tóm tắt những nguy cơ đã biết, nguy cơ tiềm ẩn và những thông tin còn thiếu của một thuốc.
Thực hiện kế hoạch Cảnh giác dược
– Mục đích của kế hoạch Cảnh giác dược là nhận biết và mô tả những nguy cơ được xác định trong phần thông tin về tính an toàn. Kế hoạch này được xây dựng nhằm:
+ Xác định mối quan ngại mới về tính an toàn;
+ Mô tả chi tiết hơn một mối quan ngại về tính an toàn đã biết, ví dụ làm sáng tỏ những yếu tố nguy cơ;
+ Điều tra xem một mối lo ngại tiềm ẩn về tính an toàn có thật hay không;
+ Cách tìm kiếm những thông tin còn thiếu.
– Hoạt động Cảnh giác dược có thể được chia thành hai loại: thực hành Cảnh giác dược thường quy và hoạt động Cảnh giác dược bổ sung.
+ Thực hành Cảnh giác dược thường quy: duy trì việc giám sát về tính an toàn và những hoạt động báo cáo theo quy định.
+ Hoạt động Cảnh giác dược bổ sung: có thể là những nghiên cứu tiền lâm sàng, thử nghiệm lâm sàng hay những nghiên cứu không can thiệp nhằm thu thập thêm thông tin về một biến cố ngoại ý được lưu ý đặc biệt; hoặc những công cụ, biện pháp đặc biệt khác để thu thập dữ liệu về biến cố bất lợi.
Lập kế hoạch cho những nghiên cứu hiệu quả hậu mãi
Mô tả những thiếu sót trong hiểu biết về hiệu quả điều trị trên nhóm người bệnh đích và sự cần thiết cần có những nghiên cứu bổ sung về hiệu quả điều trị sau khi thuốc được cấp phép lưu hành.
Lập kế hoạch giảm thiểu nguy cơ
Kế hoạch giảm thiểu nguy cơ phải cung cấp thông tin chi tiết về những biện pháp giảm thiểu nguy cơ sẽ được sử dụng để hạn chế những nguy cơ do lo ngại về tính an toàn.
Những hoạt động giảm thiểu nguy cơ có thể bao gồm những hoạt động giảm thiểu nguy cơ thường quy (ví dụ như những biện pháp liên quan đến ghi nhãn sản phẩm được cấp phép) và những hoạt động giảm thiểu nguy cơ bổ sung (ví dụ như trao đổi thông tin với nhân viên y tế, các biện pháp giáo dục, kiểm soát hệ thống phân phối). Tất cả những biện pháp giảm thiểu nguy cơ đều phải có mục tiêu rõ ràng.
– Hoạt động giảm thiểu nguy cơ thường quy là những biện pháp áp dụng cho tất cả các thuốc. Những biện pháp này liên quan đến:
+ Tóm tắt đặc tính của thuốc;
+ Nhãn thuốc;
+ Tờ rơi sản phẩm;
+ Kích thước đóng gói;
+ Tính trạng pháp lý của sản phẩm.
– Hoạt động giảm thiểu nguy cơ bổ sung là những biện pháp giảm thiểu nguy cơ không nằm trong những hoạt động thường quy kể trên. Những hoạt động giảm thiểu nguy cơ bổ sung chỉ được đề xuất khi cần thiết cho tính an toàn và hiệu quả trong sử dụng thuốc; những hoạt động này phải dựa trên căn cứ khoa học, được phát triển và đề xuất bởi người có năng lực chuyên môn phù hợp. Khi đề xuất các hoạt động giảm thiểu nguy cơ bổ sung, cần đưa ra thông tin chi tiết và lý do cần thiết phải tiến hành những hoạt động này.
Tất cả những biện pháp giảm thiểu nguy cơ phải được xem xét định kỳ và đánh giá hiệu quả.
Lập kế hoạch truyền thông nguy cơ
Việc cung cấp thông tin cho nhân viên y tế và/hoặc người bệnh về những nguy cơ cụ thể của một thuốc và những biện pháp để giảm thiểu những nguy cơ đó là một hoạt động cần thiết trong quản lý nguy cơ. Một phần quan trọng của quản lý nguy cơ là cung cấp thông tin cho những người cần thông tin đó để hiểu rõ về cân bằng nguy cơ/lợi ích của thuốc nhằm giúp họ đưa ra những quyết định sử dụng thuốc đúng đắn. Việc cung cấp những thông tin này gọi là truyền thông nguy cơ.
Truyền thông nguy cơ có thể chỉ giới hạn trong việc cung cấp thông tin trên nhãn thuốc (giảm thiểu nguy cơ thường quy) hoặc thông qua việc sử dụng những tài liệu giáo dục bổ sung. Yêu cầu về những tài liệu giáo dục bổ sung chuyên sâu hơn nội dung trên nhãn thuốc phụ thuộc vào từng nguy cơ và được xem xét trong từng trường hợp cụ thể. Bất kỳ truyền thông nguy cơ nào cũng cần nhất quán với thông tin kê đơn.
Một số ví dụ về truyền thông nguy cơ:
– Thư cập nhật thông tin dành cho nhân viên y tế;
– Cẩm nang thông tin cho người bệnh;
– Danh mục những điều cần lưu ý khi kê đơn và cấp phát thuốc;
– Các chương trình đào tạo có chủ đích (ví dụ đào tạo cấp chứng chỉ, các bài trình bày tại hội thảo khoa học);
– Xuất bản ấn phẩm khoa học.
c) Những nội dung cơ bản của Kế hoạch quản lý nguy cơ
Đơn vị kinh doanh thuốc nộp Kế hoạch quản lý nguy cơ khi có yêu cầu cụ thể của Cục Quản lý Dược dựa trên ý kiến của Hội đồng tư vấn cấp số đăng ký lưu hành thuốc Bộ Y tế. Kế hoạch quản lý nguy cơ đối với một thuốc bao gồm các nội dung:
Tổng quan về thuốc
Cung cấp những thông tin liên quan đến việc quản lý đối với Kế hoạch quản lý nguy cơ và thông tin tóm tắt về (các) sản phẩm liên quan.
Thông tin về tính an toàn
Tổng hợp những dữ liệu hiện có về tính an toàn, bao gồm những dữ liệu nghiên cứu tiền lâm sàng và lâm sàng, các nghiên cứu dịch tễ; mô tả những nguy cơ đã biết và nguy cơ tiềm ẩn; xác định nhóm người bệnh có khả năng gặp nguy cơ và xác định những nghi vấn nổi bật về tính an toàn cần phải có những đánh giá sâu hơn.
Kế hoạch Cảnh giác dược
Mô tả những hoạt động Cảnh giác dược nhằm mục đích mô tả rõ hơn những nguy cơ đã biết và nguy cơ tiềm ẩn.
Kế hoạch cho những nghiên cứu hiệu quả hậu mãi
Mô tả những thiếu sót nếu có trong hiểu biết về hiệu quả trên nhóm người bệnh đích và yêu cầu đối với những nghiên cứu hậu mãi sâu hơn về hiệu quả điều trị.
Các biện pháp giảm thiểu nguy cơ
Trong trường hợp cần thiết có những hoạt động giảm thiểu nguy cơ bổ sung ngoài nhãn thuốc, kế hoạch giảm thiểu nguy cơ phải được xây dựng với đầy đủ chi tiết về những hoạt động nhằm hạn chế những nguy cơ liên quan.
Tổng kết Kế hoạch quản lý nguy cơ
Tóm tắt những yếu tố chủ chốt trong Kế hoạch quản lý nguy cơ với trọng tâm là các hoạt động giảm thiểu nguy cơ.
Ngoài các nội dung trên, Cục Quản lý Dược có thể yêu cầu thêm phần phụ lục gồm các thông tin:
– Tóm tắt các đặc tính sản phẩm, tờ hướng dẫn sử dụng thuốc, tờ thông tin cho người bệnh;
– Tình trạng lưu hành trên thế giới;
– Các nghiên cứu lâm sàng đã hoàn tất;
– Các báo cáo nghiên cứu mới;
– Các thông tin hỗ trợ khác.
Lưu ý: đơn vị kinh doanh thuốc có thể nộp Kế hoạch quản lý nguy cơ theo mẫu của doanh nghiệp với điều kiện Kế hoạch quản lý nguy cơ đó chứa đầy đủ nội dung theo yêu cầu của cơ quan quản lý. Doanh nghiệp có thể xây dựng Kế hoạch quản lý nguy cơ mới hoặc đệ trình Kế hoạch quản lý nguy cơ được áp dụng ở các nước khác tại thời điểm được phê duyệt (có thể bao gồm những cập nhật, bổ sung) cùng phụ lục liên quan đến những vấn đề riêng tại Việt Nam trong đó mô tả Kế hoạch Cảnh giác dược và Kế hoạch giảm thiểu nguy cơ áp dụng riêng cho Việt Nam so với nội dung đã nêu trong bản Kế hoạch quản lý nguy cơ được áp dụng tại các nước khác.
d) Những yêu cầu về Kế hoạch quản lý nguy cơ ở Việt Nam
Phạm vi áp dụng
Khi thuốc được cơ quan quản lý yêu cầu cần giám sát về tính an toàn.
Thời hạn đệ trình
Thời hạn này tùy thuộc tính chất và mức độ an toàn của thuốc và do cơ quan quản lý quyết định.
Thực hiện và lưu trữ
Khi Kế hoạch quản lý nguy cơ đã được thông qua, đơn vị kinh doanh thuốc phải áp dụng Kế hoạch quản lý nguy cơ theo đúng trách nhiệm và lưu trữ tài liệu liên quan đến việc áp dụng Kế hoạch này để sẵn sàng cung cấp khi có yêu cầu thanh tra/kiểm tra.
Ngôn ngữ
Ngôn ngữ được chấp nhận sử dụng trong Kế hoạch quản lý nguy cơ là tiếng Việt và/hoặc tiếng Anh.
Cơ quan tiếp nhận Kế hoạch quản lý nguy cơ:
Kế hoạch quản lý nguy cơ phải được gửi chính thức qua đường bưu điện đến Cục Quản lý Dược – Bộ Y tế theo địa chỉ ở mục 6.2.2 của tài liệu này.
B. Hoạt động Cảnh giác dược tại các cơ sở bán lẻ thuốc
6.3. Các nhà thuốc đạt tiêu chuẩn thực hành tốt nhà thuốc (GPP)
Các nhà thuốc đạt tiêu chuẩn thực hành tốt nhà thuốc cần theo dõi và thông báo về các tác dụng không mong muốn của thuốc theo quy định về nguyên tắc, tiêu chuẩn thực hành tốt nhà thuốc.
– Báo cáo các biến cố bất lợi do người mua thuốc phản ánh theo mẫu báo cáo phản ứng có hại của thuốc (xem phụ lục 1 của Hướng dẫn này).
– Khuyến khích báo cáo các vấn đề liên quan đến chất lượng thuốc về cảm quan được cơ sở phát hiện (xem phụ lục 7 của Hướng dẫn này).
Báo cáo gửi về một trong hai địa chỉ đã nêu tại mục 6.2.1.
6.4. Các cơ sở bán lẻ khác
– Ghi nhận phản ánh của người mua thuốc đối với các biến cố bất lợi xảy ra khi dùng thuốc.
– Báo cáo các biến cố bất lợi do người mua thuốc phản ánh theo mẫu báo cáo phản ứng có hại của thuốc (xem phụ lục 1 của Hướng dẫn này).
– Khuyến khích các cơ sở báo cáo các vấn đề liên quan đến chất lượng thuốc về mặt cảm quan được cơ sở phát hiện (xem phụ lục 7 của Hướng dẫn này).
Báo cáo gửi về một trong hai địa chỉ nêu tại mục 6.2.1 của tài liệu này.
CHƯƠNG 7.
THEO DÕI BIẾN CỐ BẤT LỢI CỦA THUỐC TRONG THỬ NGHIỆM LÂM SÀNG
7.1. Nguyên tắc chung
Trong quá trình nghiên cứu thử nghiệm lâm sàng, việc ghi nhận và báo cáo các biến cố bất lợi xảy ra trong quá trình nghiên cứu là phần quan trọng thiết yếu. Một trong các mục tiêu của nghiên cứu lâm sàng là xác định tính an toàn của sản phẩm thông qua kết quả nghiên cứu. Mục tiêu này được thực hiện bằng việc ghi chép, báo cáo và phân tích các biến cố bất lợi trong quá trình nghiên cứu. Các dữ liệu về tính an toàn quan trọng đối với quy trình đăng ký cấp phép lưu hành sản phẩm mới. Ngoài ra, về mặt đạo đức, các báo cáo biến cố bất lợi sẽ giúp ích cho việc bảo vệ an toàn cho đối tượng nghiên cứu và tính toàn vẹn của dữ liệu nghiên cứu.
Việc ghi nhận và báo cáo các biến cố bất lợi đầy đủ nhằm thu thập dữ liệu an toàn một cách hệ thống, đồng thời bổ sung thông tin về tính an toàn của sản phẩm nghiên cứu. Mặt khác, việc báo cáo các biến cố bất lợi giúp cho việc cảnh báo cho nhà tài trợ, cơ quan quản lý và hội đồng đạo đức các nguy cơ về an toàn cho đối tượng nghiên cứu. Thêm vào đó, báo cáo biến cố bất lợi còn hỗ trợ cho các kế hoạch giám sát, kiểm tra nghiên cứu.
Việc xử trí, ghi nhận, báo cáo các biến cố bất lợi, biến cố bất lợi nghiêm trọng trong các thử nghiệm lâm sàng tiến hành tại Việt Nam cần tuân thủ theo các hướng dẫn về Thực hành lâm sàng tốt (Good Clinical Practice, GCP) quốc tế và Việt Nam.
Hướng dẫn này áp dụng cho các thử nghiệm lâm sàng thuốc, bao gồm: thử nghiệm lâm sàng nhằm chứng minh tính an toàn hoặc hiệu lực/hiệu quả của thuốc trong phát triển thuốc mới và phương pháp điều trị mới, các nghiên cứu trên người về dược động học, sinh khả dụng và tương đương sinh học của thuốc.
7.2. Trách nhiệm của các bên trong việc theo dõi và báo cáo biến cố bất lợi trong thử nghiệm lâm sàng tiến hành tại Việt Nam
7.2.1. Nghiên cứu viên chính, nghiên cứu viên tại điểm nghiên cứu
Nghiên cứu viên chính, nghiên cứu viên tại điểm nghiên cứu chịu trách nhiệm về việc phát hiện, xử trí AE/SAE đảm bảo kịp thời, an toàn cho đối tượng nghiên cứu; theo dõi và ghi nhận đầy đủ các thông tin; gửi báo cáo AE/SAE cho nhà tài trợ, Hội đồng Đạo đức cấp cơ sở, Ban Đánh giá vấn đề đạo đức trong nghiên cứu y sinh học Bộ Y tế. Trong trường hợp mức độ và tần suất AE/SAE vượt quá giới hạn cho phép, nghiên cứu viên có thể đề xuất với nhà tài trợ và Hội đồng Đạo đức trong nghiên cứu y sinh học tạm dừng thử nghiệm.
7.2.2. Tổ chức nhận thử, đơn vị triển khai nghiên cứu
Tổ chức nhận thử, đơn vị triển khai nghiên cứu chịu trách nhiệm quản lý, giám sát việc phát hiện, xử trí, theo dõi báo cáo AE/ SAE tại điểm nghiên cứu đảm bảo an toàn cho đối tượng nghiên cứu.
7.2.3. Hội đồng Đạo đức/Khoa học cấp cơ sở của tổ chức nhận thử
Hội đồng Đạo đức/Khoa học cấp cơ sở của tổ chức nhận thử xem xét cho ý kiến chuyên môn về các AE/SAE xảy ra tại điểm nghiên cứu, đảm bảo an toàn cho đối tượng nghiên cứu.
7.2.4. Nhà tài trợ và các tổ chức được nhà tài trợ ủy quyền
Nhà tài trợ và các tổ chức được nhà tài trợ ủy quyền (tổ chức, cá nhân có thuốc sản phẩm thử nghiệm lâm sàng; tổ chức nghiên cứu hợp đồng, tổ chức giám sát địa điểm nghiên cứu) chịu trách nhiệm:
– Phối hợp với nghiên cứu viên chính báo cáo AE/SAE xảy ra tại các điểm nghiên cứu tại Việt Nam gửi về Hội đồng đạo đức cấp cơ sở của tổ chức nhận thử/đơn vị chủ trì và Văn phòng Ban Đánh giá vấn đề đạo đức trong nghiên cứu y sinh học Bộ Y tế;
– Cập nhật các thông tin về ADR ngoài dự kiến của sản phẩm nghiên cứu tại các điểm nghiên cứu để thông báo cho các nghiên cứu viên và bổ sung vào Hồ sơ sản phẩm nghiên cứu;
– Báo cáo các Phản ứng bất lợi nghiêm trọng ngoài dự kiến (SUSAR) về Cục Khoa học công nghệ và Đào tạo – Bộ Y tế;
– Tổng hợp dữ liệu các biến cố bất lợi, biến cố bất lợi nghiêm trọng đưa vào báo cáo tiến độ định kỳ hàng năm và báo cáo tổng kết kết quả nghiên cứu.
7.2.5. Ban Đánh giá vấn đề đạo đức trong nghiên cứu y sinh học Bộ Y tế
Ban Đánh giá vấn đề đạo đức trong nghiên cứu y sinh học Bộ Y tế chịu trách nhiệm xem xét, đánh giá các báo cáo SAE, báo cáo định kỳ hàng năm và phản hồi cho nghiên cứu viên bằng văn bản. Trong trường hợp cần thiết, tổ chức giám sát kiểm tra điểm nghiên cứu và tư vấn cho cơ quan quản lý để có chỉ đạo kịp thời đối với Nghiên cứu viên, tổ chức nhận thử/Đơn vị triển khai nghiên cứu, nhà tài trợ nhằm đảm bảo an toàn cho đối tượng nghiên cứu.
7.2.6. Trung tâm Quốc gia về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc
Trung tâm Quốc gia về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc có trách nhiệm phối hợp với Ban Đánh giá vấn đề đạo đức trong nghiên cứu y sinh học Bộ Y tế để phân tích, thống kê dữ liệu các báo cáo AE/SAE trong các thử nghiệm lâm sàng.
7.3. Quy trình, thời hạn và biểu mẫu báo cáo
7.3.1. Báo cáo khẩn cấp
Đối với tất cả các SAE: nghiên cứu viên chính có trách nhiệm báo cáo khẩn cấp cho nhà tài trợ và Hội đồng Đạo đức cấp cơ sở của tổ chức nhận thử trong thời gian 24 giờ kể từ khi được biết thông tin. Tùy theo từng loại SAE, việc báo cáo cho Ban Đánh giá vấn đề đạo đức trong nghiên cứu y sinh học Bộ Y tế và các tổ chức liên quan như sau:
a) Đối với các SAE gây tử vong hoặc đe dọa tính mạng
Nghiên cứu viên chính phối hợp với nhà tài trợ hoàn thiện thông tin và gửi báo cáo về Văn phòng Ban Đánh giá vấn đề đạo đức trong nghiên cứu y sinh học Bộ Y tế. Báo cáo bằng văn bản gửi trong thời gian sớm nhất có thể nhưng không muộn hơn 7 ngày theo lịch kể từ khi có thông tin SAE. Nội dung báo cáo ban đầu theo Biểu mẫu báo cáo SAE (xem phụ lục 32 của Hướng dẫn này) nhưng không nhất thiết phải đầy đủ thông tin tại thời điểm báo cáo. Nếu báo cáo ban đầu chưa đầy đủ thông tin, cần có báo cáo theo dõi tiếp theo đầy đủ chi tiết các phần của biểu mẫu báo cáo, được hoàn tất và gửi trong vòng 15 ngày theo lịch kể từ thời điểm có thông tin SAE. Nếu có thông tin bổ sung về SAE, cần báo cáo theo dõi tiếp theo đầy đủ thông tin trong vòng 7 ngày theo lịch kể từ khi có thông tin bổ sung.
b) Đối với các SAE không thuộc loại gây tử vong hoặc đe dọa đến tính mạng
Nghiên cứu viên chính phối hợp với nhà tài trợ hoàn thiện thông tin và gửi báo cáo SAE chi tiết về Ban Đánh giá vấn đề đạo đức trong nghiên cứu y sinh học Bộ Y tế trong thời gian sớm nhất nhưng không muộn hơn 15 ngày theo lịch kể từ thời điểm có thông tin SAE.
c) Đối với các phản ứng bất lợi nghiêm trọng ngoài dự kiến
Trong trường hợp nhà tài trợ xác định SAE là phản ứng bất lợi nghiêm trọng ngoài dự kiến (SUSAR), nhà tài trợ gửi công văn báo cáo kèm theo biểu mẫu CIOMS (xem phụ lục 28 của hướng dẫn này) về Cục Khoa học công nghệ và Đào tạo, Bộ Y tế trong vòng 15 ngày theo lịch kể từ khi nhà tài trợ xác định là SUSAR.
7.3.2. Báo cáo định kỳ
Hàng năm, nhà tài trợ phải gửi báo cáo định kỳ của từng nghiên cứu thử nghiệm lâm sàng đang tiến hành tại Việt Nam về Ban Đánh giá vấn đề đạo đức trong nghiên cứu y sinh học Bộ Y tế. Trong nội dung báo cáo định kỳ hàng năm của từng nghiên cứu phải có các nội dung liên quan đến tính an toàn, bao gồm các thông tin tổng hợp về biến cố bất lợi và biến cố bất lợi nghiêm trọng, số lượng tích lũy tính đến ngày báo cáo, theo biểu mẫu báo cáo định kỳ (xem phụ lục 33 của Hướng dẫn này) kèm theo danh sách các SAE xảy ra tại các điểm nghiên cứu ở Việt Nam. Đối với các nghiên cứu đa trung tâm, đa quốc gia có thể bổ sung thêm tình hình các AE/SAE chung của cả nghiên cứu.
Thời hạn gửi báo cáo định kỳ hàng năm tính từ thời điểm nghiên cứu được phê duyệt đến ngày báo cáo là 12 tháng. Báo cáo định kỳ năm tiếp theo được tính từ thời điểm phải báo cáo của năm trước.
7.3.3. Nơi nhận báo cáo
Các báo cáo về biến cố bất lợi trong các thử nghiệm lâm sàng tại Việt Nam phải được báo cáo về Văn phòng Ban Đánh giá vấn đề đạo đức trong nghiên cứu y sinh học Bộ Y tế. Trung tâm Quốc gia về Thông tin thuốc và Theo dõi phản ứng có hại của thuốc cùng phối hợp để phân tích xử lý thông tin dữ liệu báo cáo.
7.3.4. Hoạt động của các cơ quan liên quan đối với báo cáo AE/ SAE
Trên cơ sở các báo cáo AE/SAE, tùy theo tính chất nghiêm trọng của các AE/SAE và mức độ liên quan đến sản phẩm nghiên cứu, các cơ quan có liên quan (nhà tài trợ, Hội đồng đạo đức trong nghiên cứu y sinh học các cấp, cơ quan quản lý) sau khi đánh giá các báo cáo có thể có những hành động sau:
a) Yêu cầu nghiên cứu viên bổ sung các thông tin liên quan đến AE/SAE được báo cáo.
b) Tạm dừng nghiên cứu để xem xét lại tiêu chuẩn tuyển chọn đối tượng, quy trình tuyển chọn đối tượng và tính hợp lý của việc tiếp tục triển khai nghiên cứu.
c) Điều chỉnh tiêu chí tuyển chọn và quy trình tuyển chọn đối tượng, tiếp tục triển khai nghiên cứu sau khi đã tạm dừng nghiên cứu và xem xét các vấn đề liên quan.
d) Chấm dứt nghiên cứu nếu AE/SAE xảy ra ở tần suất, mức độ cao, thể hiện sản phẩm nghiên cứu không an toàn cho việc sử dụng trên người.
e) Thực hiện kiểm tra, thanh tra điểm nghiên cứu về các AE/ SAE để có thể đưa ra các hành động nêu trên.
Trong trường hợp các AE/SAE được xem xét đánh giá là không gây những rủi ro nghiêm trọng ảnh hưởng đến sự an toàn của đối tượng nghiên cứu và không liên quan đến thuốc nghiên cứu, nghiên cứu vẫn tiếp tục được tiến hành.
CHƯƠNG 8.
THÔNG TIN THUỐC TRONG HOẠT ĐỘNG CẢNH GIÁC DƯỢC
8.1. Vai trò của các nguồn dữ liệu tra cứu thông tin thuốc trong hoạt động Cảnh giác dược
Thông tin thuốc có ý nghĩa quan trọng trong việc hướng dẫn lựa chọn, sử dụng thuốc hợp lý, an toàn. Với sự lớn mạnh và tính phổ biến của công nghệ thông tin, thông tin thuốc đang có sự phát triển cả về số lượng cũng như chiều sâu. Rất nhiều nguồn dữ liệu thông tin thuốc khác nhau đã ra đời phục vụ công tác tra cứu trong thực hành lâm sàng. Các cơ sở dữ liệu này có vai trò lưu trữ và cập nhật các thông tin về nhiều lĩnh vực, bao gồm cả an toàn thuốc. Thông tin trong các cơ sở dữ liệu là cơ sở để nhân viên y tế nhận định phản ứng có hại xảy ra. Việc đánh giá kịp thời, chính xác thông tin về phản ứng có hại của thuốc, các biện pháp dự phòng và xử trí có ảnh hưởng tích cực đến kết quả điều trị của người bệnh.
Đồng thời, thông tin về an toàn thuốc liên tục được cập nhật dựa trên kết quả các nghiên cứu lâm sàng cũng như qua việc giám sát thuốc sau khi được đăng ký lưu hành trên thị trường giúp cho các nhân viên y tế nắm bắt các thông tin mới nhất về an toàn thuốc. Các thông tin này là cơ sở để nhân viên y tế đưa ra các quyết định sử dụng thuốc phù hợp với thực tế lâm sàng tại cơ sở điều trị. Nhiệm vụ tư vấn cho thầy thuốc trong việc kê đơn và điều trị này của đơn vị thông tin thuốc đã được đề cập trong Công văn số 10766/YT-ĐTr ngày 13/11/2003 của Vụ Điều trị (nay là Cục Quản lý Khám, chữa bệnh). Thông tư số 13/2009/TT-BYT ban hành ngày 01/09/2009 cũng đã nêu rõ nhiệm vụ cung cấp thông tin thuốc nhằm đảm bảo sử dụng thuốc an toàn, hợp lý trong phạm vi bệnh viện của đơn vị thông tin thuốc. Công việc này sau đó đã được xác định là một trong những nhiệm vụ chuyên môn của dược sĩ lâm sàng, quy định tại Thông tư số 31/2012/TT-BYT ngày 20/12/2012 của Bộ trưởng Bộ Y tế về hướng dẫn hoạt động dược lâm sàng trong bệnh viện. Với vai trò như là cầu nối đưa thông tin, dược sĩ lâm sàng có trách nhiệm cập nhật thông tin sử dụng thuốc, thông tin về thuốc mới, thông tin Cảnh giác dược gửi đến nhân viên y tế và đến người bệnh bằng nhiều hình thức khác nhau như: trực tiếp, văn bản, bảng tin bệnh viện, thư điện tử, tranh ảnh, tờ hướng dẫn, trang thông tin điện tử.
8.2. Cập nhật thông tin về an toàn thuốc
8.2.1. Quy trình cập nhật và truyền thông về an toàn thuốc
Quy trình cập nhật và truyền thông về an toàn thuốc tại cơ sở khám bệnh, chữa bệnh được mô tả trong hình 6. Các chương trình y tế quốc gia, các đơn vị kinh doanh thuốc và các nhà quản lý cũng có thể sử dụng các nguồn tài liệu cập nhật thông tin thuốc trong hoạt động Cảnh giác dược tại cơ sở của mình.
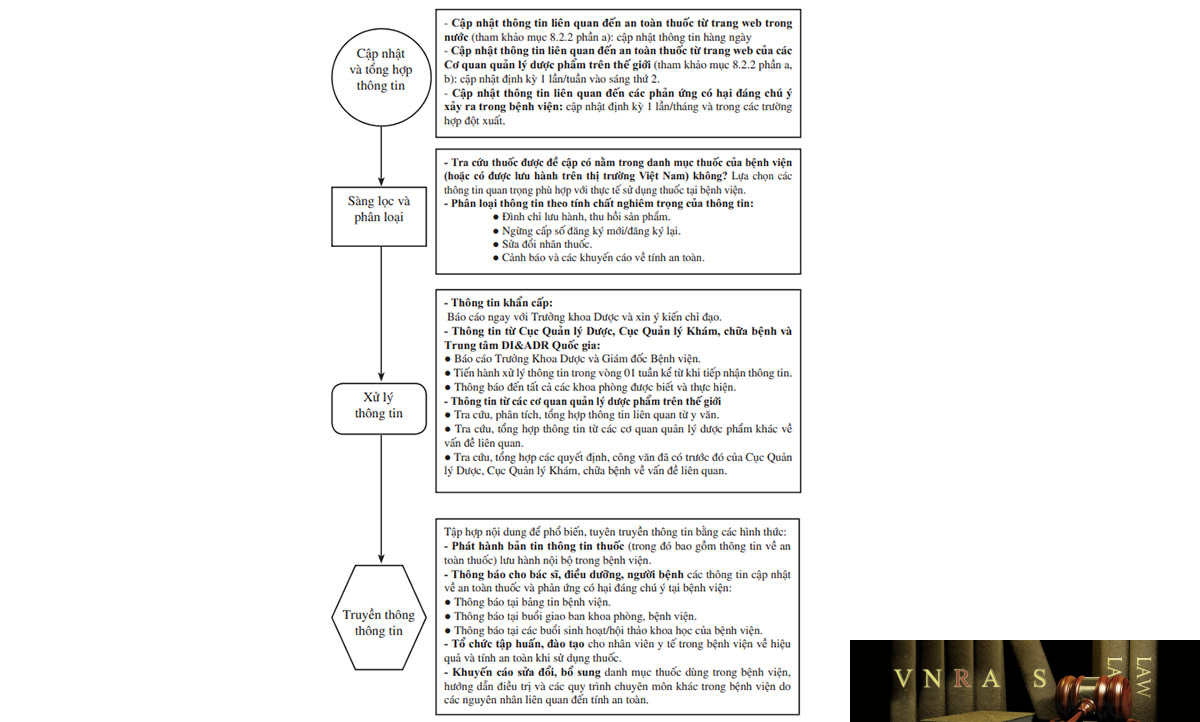
8.2.2. Các nguồn tài liệu cập nhật thông tin thuốc
a) Trang web của một số Cơ quan Quản lý Dược phẩm trên thế giới và tại Việt Nam
Trang web của các cơ quan quản lý dược phẩm trên thế giới và tại Việt Nam liên tục được cập nhật các thông tin về an toàn thuốc. Đây là nguồn dữ liệu quan trọng trong thực hành lâm sàng. Địa chỉ và cách thức truy cập các trang web của một số cơ quan quản lý dược phẩm trên thế giới và tại Việt Nam được trình bày trong bảng 3.
VĂN BẢN DẠNG WORD: 2111_QD_BYT_VNRAS
Hướng dẫn quốc gia về cảnh giác Dược












{kind=link}